

Noonan syndrome: clinical aspects and molecular pathogenesis
- PMID: 20648242
- PMCID: PMC2858523
- DOI: 10.1159/000276766
Noonan syndrome: clinical aspects and molecular pathogenesis
Abstract
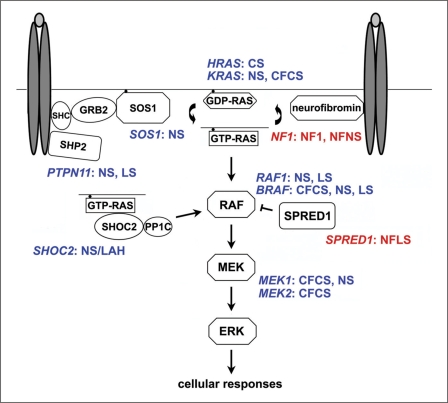
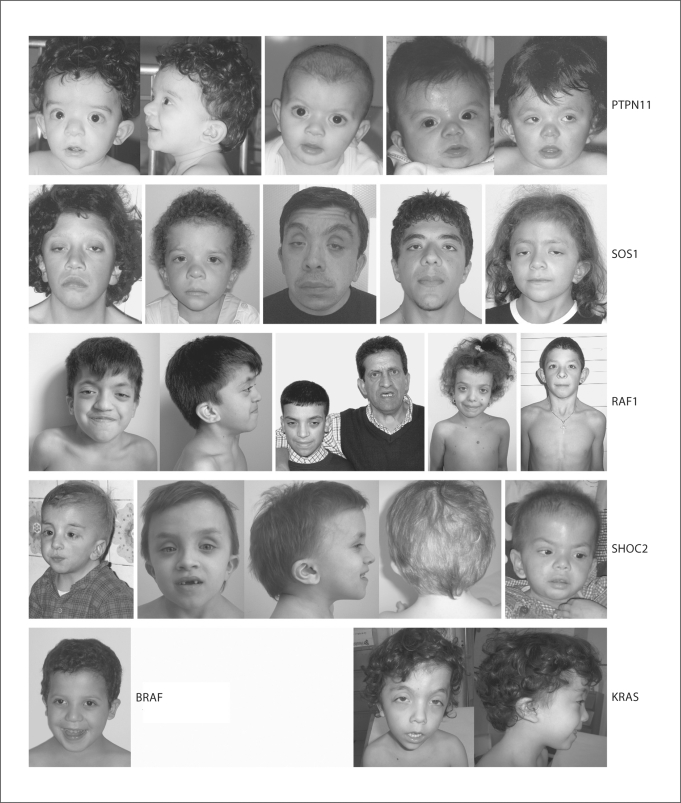
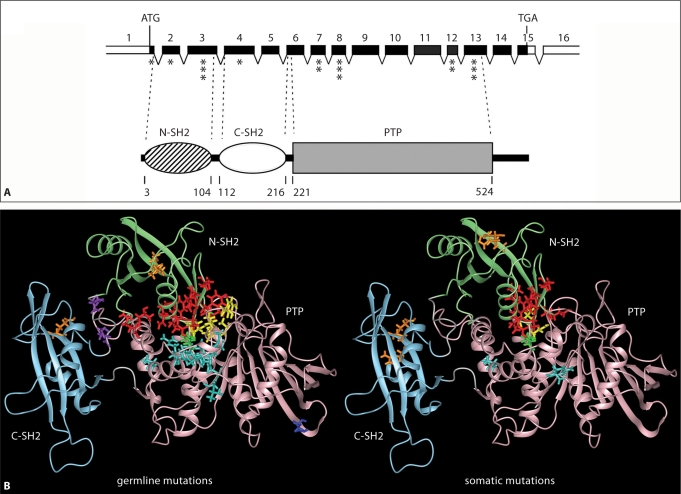
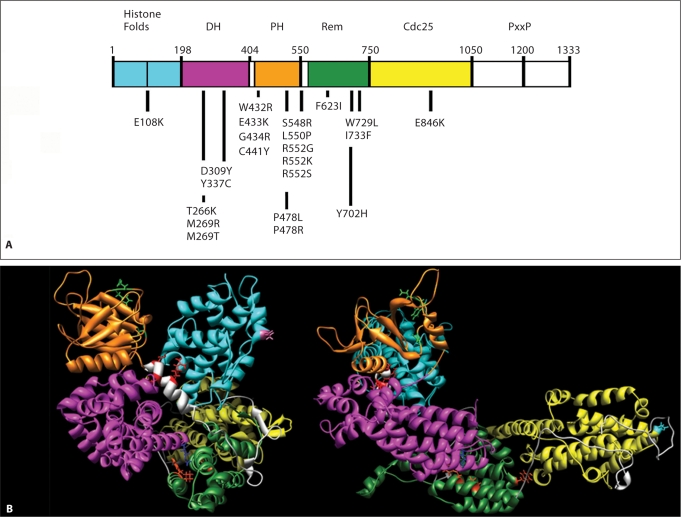
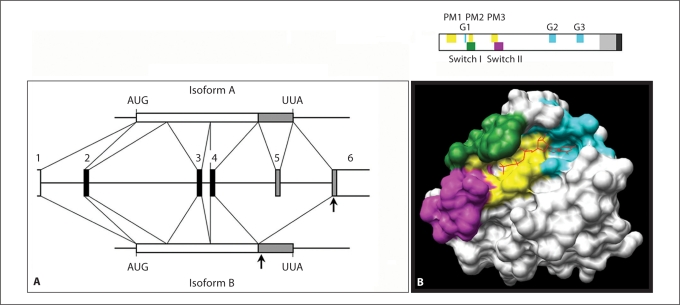
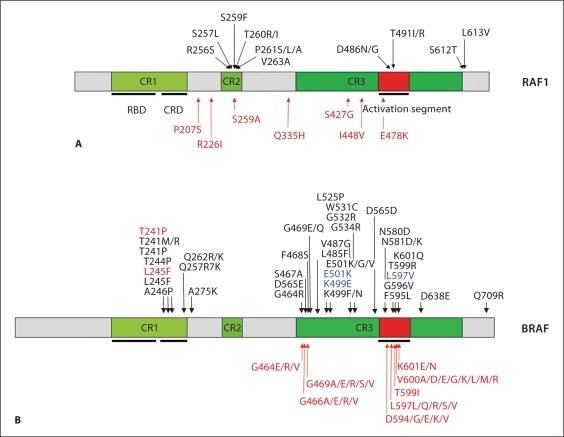
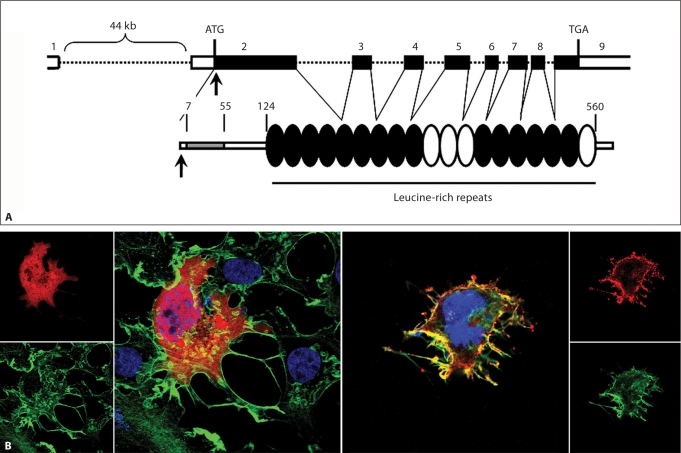
Noonan syndrome (NS) is a relatively common, clinically variable and genetically heterogeneous developmental disorder characterized by postnatally reduced growth, distinctive facial dysmorphism, cardiac defects and variable cognitive deficits. Other associated features include ectodermal and skeletal defects, cryptorchidism, lymphatic dysplasias, bleeding tendency, and, rarely, predisposition to hematologic malignancies during childhood. NS is caused by mutations in the PTPN11, SOS1, KRAS, RAF1, BRAF and MEK1 (MAP2K1) genes, accounting for approximately 70% of affected individuals. SHP2 (encoded by PTPN11), SOS1, BRAF, RAF1 and MEK1 positively contribute to RAS-MAPK signaling, and possess complex autoinhibitory mechanisms that are impaired by mutations. Similarly, reduced GTPase activity or increased guanine nucleotide release underlie the aberrant signal flow through the MAPK cascade promoted by most KRAS mutations. More recently, a single missense mutation in SHOC2, which encodes a cytoplasmic scaffold positively controlling RAF1 activation, has been discovered to cause a closely related phenotype previously termed Noonan-like syndrome with loose anagen hair. This mutation promotes aberrantly acquired N-myristoylation of the protein, resulting in its constitutive targeting to the plasma membrane and dysregulated function. PTPN11, BRAF and RAF1 mutations also account for approximately 95% of LEOPARD syndrome, a condition which resembles NS phenotypically but is characterized by multiple lentigines dispersed throughout the body, café-au-lait spots, and a higher prevalence of electrocardiographic conduction abnormalities, obstructive cardiomyopathy and sensorineural hearing deficits. These recent discoveries demonstrate that the substantial phenotypic variation characterizing NS and related conditions can be ascribed, in part, to the gene mutated and even the specific molecular lesion involved.
Keywords: Cardiofaciocutaneous syndrome; Costello syndrome; Genotype-phenotype correlations; LEOPARD syndrome; Molecular basis of disease; Molecular epidemiology; Mutation analysis; Neurocardiofacialcutaneous syndrome family; Noonan syndrome; RAS signaling.
Figures
References
-
- Adekunle O, Gopee A, el-Sayed M, Thilaganathan B. Increased first trimester nuchal translucency: Pregnancy and infant outcomes after routine screening for Down's syndrome in an unselected antenatal population. Br J Radiol. 1999;72:457–460. - PubMed
-
- Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–1074. - PubMed
-
- Ahmed ML, Foot AB, Edge JA, Lamkin VA, Savage MO, Dunger DB. Noonan's syndrome: Abnormalities of the growth hormone/IGF axis and the response to treatment with human biosynthetic growth hormone. Acta Paediatr Scand. 1991;80:446–450. - PubMed
-
- Alfieri P, Cesarini L, Zampino G, Pantaleoni F, Selicorni A, et al. Visual function in Noonan and LEOPARD syndrome. Neuropediatrics. 2008;39:335–340. - PubMed
-
- Allanson JE. Noonan syndrome. Am J Med Genet C Semin Med Genet. 2007;145C:274–279. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
