

Targets for cystic fibrosis therapy: proteomic analysis and correction of mutant cystic fibrosis transmembrane conductance regulator
- PMID: 20653506
- PMCID: PMC2927865
- DOI: 10.1586/epr.10.45
Targets for cystic fibrosis therapy: proteomic analysis and correction of mutant cystic fibrosis transmembrane conductance regulator
Abstract
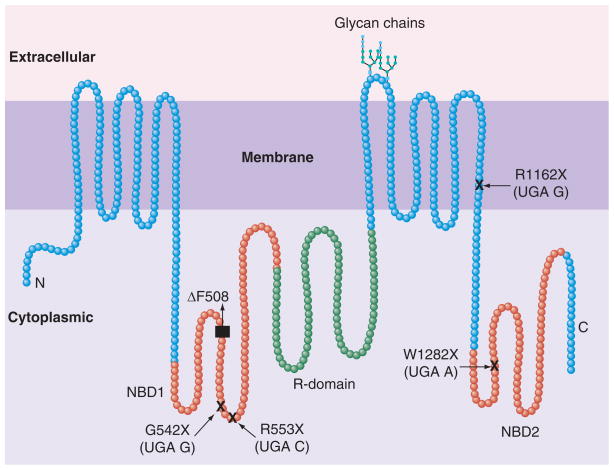
Proteomic analysis has proved to be an important tool for understanding the complex nature of genetic disorders, such as cystic fibrosis (CF), by defining the cellular protein environment (proteome) associated with wild-type and mutant proteins. Proteomic screens identified the proteome of CF transmembrane conductance regulator (CFTR), and provided fundamental information to studies designed for understanding the crucial components of physiological CFTR function. Simultaneously, high-throughput screens for small-molecular correctors of CFTR mutants provided promising candidates for therapy. The majority of CF cases are caused by nucleotide deletions (DeltaF508 CFTR; >75%), resulting in CFTR misfolding, or insertion of premature termination codons ( approximately 10%), leading to unstable mRNA and reduced levels of truncated dysfunctional CFTR. In this article, we review recent results of proteomic screens, developments in identifying correctors for the most frequent CFTR mutants, and comment on how integration of the knowledge gained from these studies may aid in finding a cure for CF and a number of other genetic disorders.
Figures
References
-
- Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. - PubMed
-
- Quinton PM. Chloride impermeability in cystic fibrosis. Nature. 1983;301(5899):421–422. - PubMed
-
- Quinton PM, Bijman J. Higher bioelectric potentials due to decreased chloride absorption in the sweat glands of patients with cystic fibrosis. N Engl J Med. 1983;308(20):1185–1189. - PubMed
-
- Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. - PubMed
-
- Griesenbach U, Alton EW. Cystic fibrosis gene therapy: successes, failures and hopes for the future. Expert Rev Respir Med. 2009;3(4):363–371. - PubMed
Website
-
- Cystic Fibrosis Mutation Database. www.genet.sickkids.on.ca/cftr.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical