

Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability
- PMID: 20655035
- PMCID: PMC2917707
- DOI: 10.1016/j.ajhg.2010.06.017
Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability
Abstract
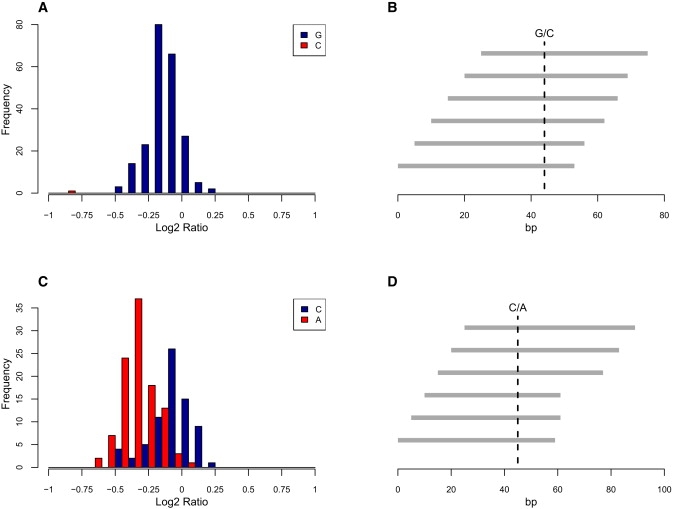
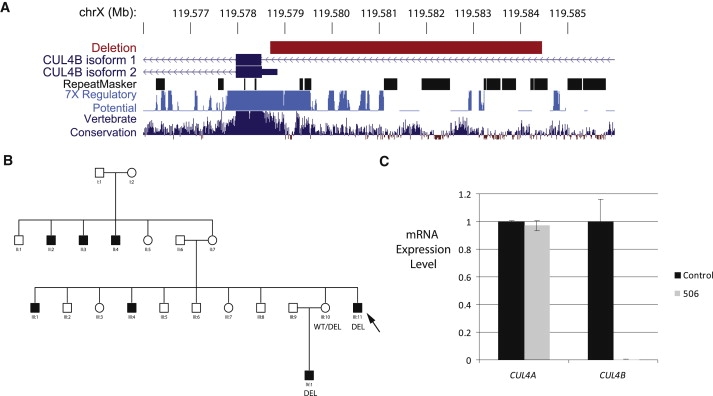
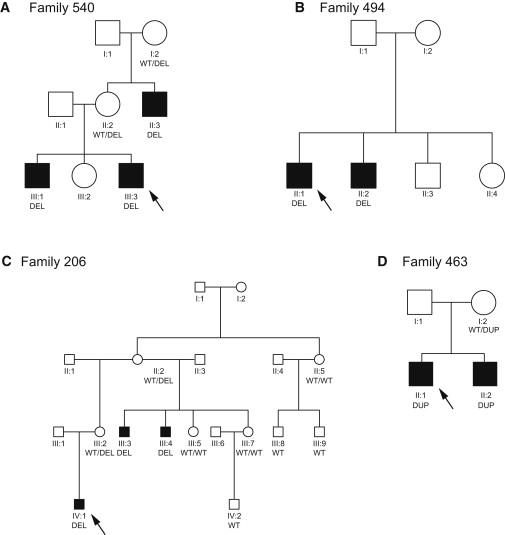
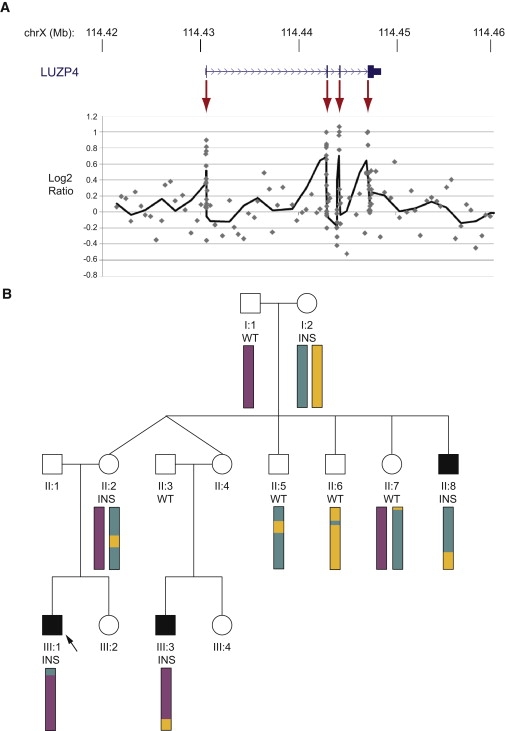
Copy number variants and indels in 251 families with evidence of X-linked intellectual disability (XLID) were investigated by array comparative genomic hybridization on a high-density oligonucleotide X chromosome array platform. We identified pathogenic copy number variants in 10% of families, with mutations ranging from 2 kb to 11 Mb in size. The challenge of assessing causality was facilitated by prior knowledge of XLID-associated genes and the ability to test for cosegregation of variants with disease through extended pedigrees. Fine-scale analysis of rare variants in XLID families leads us to propose four additional genes, PTCHD1, WDR13, FAAH2, and GSPT2, as candidates for XLID causation and the identification of further deletions and duplications affecting X chromosome genes but without apparent disease consequences. Breakpoints of pathogenic variants were characterized to provide insight into the underlying mutational mechanisms and indicated a predominance of mitotic rather than meiotic events. By effectively bridging the gap between karyotype-level investigations and X chromosome exon resequencing, this study informs discussion of alternative mutational mechanisms, such as noncoding variants and non-X-linked disease, which might explain the shortfall of mutation yield in the well-characterized International Genetics of Learning Disability (IGOLD) cohort, where currently disease remains unexplained in two-thirds of families.
Figures
References
-
- World Health Organization; Geneva: 1992. The ICD-10 Classification of Mental and Behavioural Disorders.
-
- American Psychiatric Association; Washington, D.C.: 1994. Diagnostic and Statistical Manual of Mental Disorders DSM-IV.
-
- Leonard H., Wen X. The epidemiology of mental retardation: Challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 2002;8:117–134. - PubMed
-
- Ropers H.H. Genetics of intellectual disability. Curr. Opin. Genet. Dev. 2008;18:241–250. - PubMed
-
- Schreppers-Tijdink G.A., Curfs L.M., Wiegers A., Kleczkowska A., Fryns J.P. A systematic cytogenetic study of a population of 1170 mentally retarded and/or behaviourly disturbed patients including fragile X-screening. The Hondsberg experience. J. Genet. Hum. 1988;36:425–446. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
