

Epidemic history and evolutionary dynamics of hepatitis B virus infection in two remote communities in rural Nigeria
- PMID: 20657838
- PMCID: PMC2906510
- DOI: 10.1371/journal.pone.0011615
Epidemic history and evolutionary dynamics of hepatitis B virus infection in two remote communities in rural Nigeria
Abstract
Background: In Nigeria, hepatitis B virus (HBV) infection has reached hyperendemic levels and its nature and origin have been described as a puzzle. In this study, we investigated the molecular epidemiology and epidemic history of HBV infection in two semi-isolated rural communities in North/Central Nigeria. It was expected that only a few, if any, HBV strains could have been introduced and effectively transmitted among these residents, reflecting limited contacts of these communities with the general population in the country.
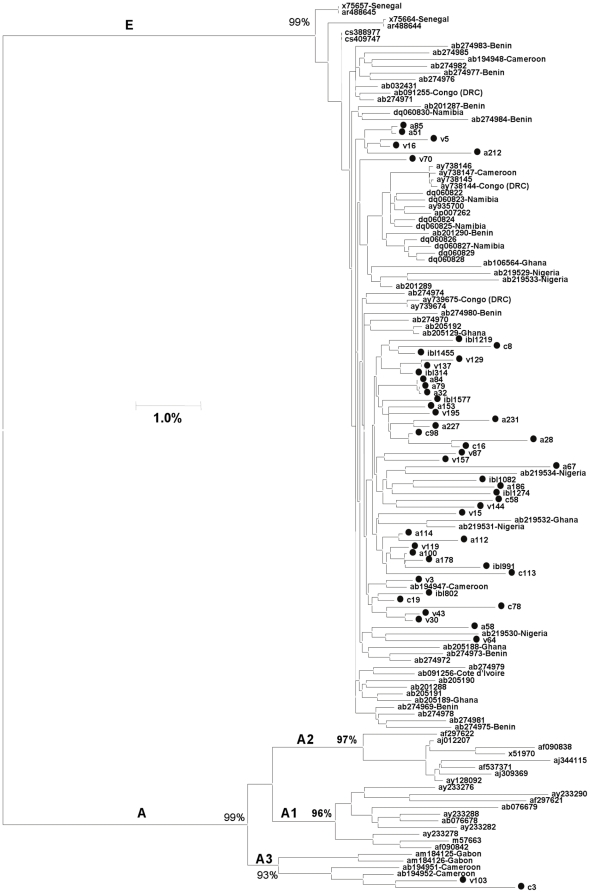
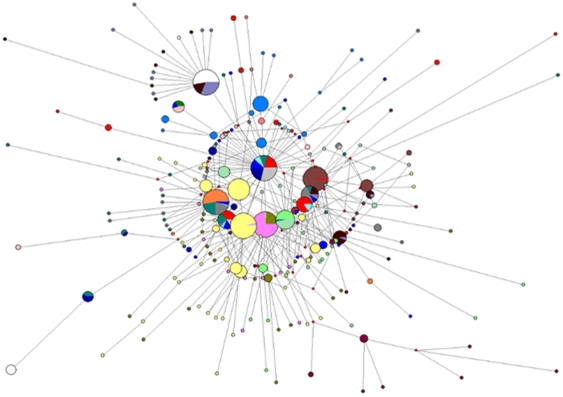
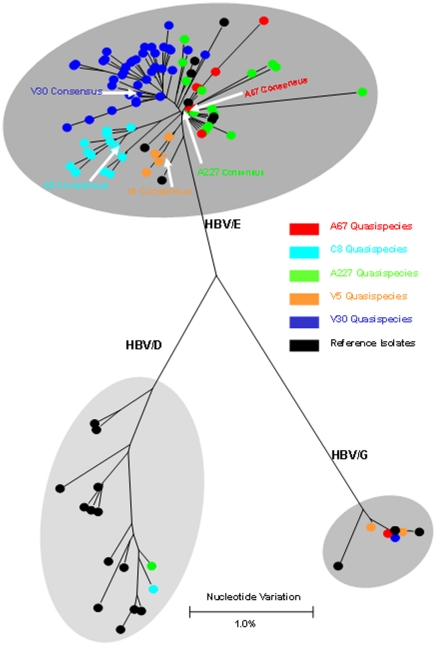
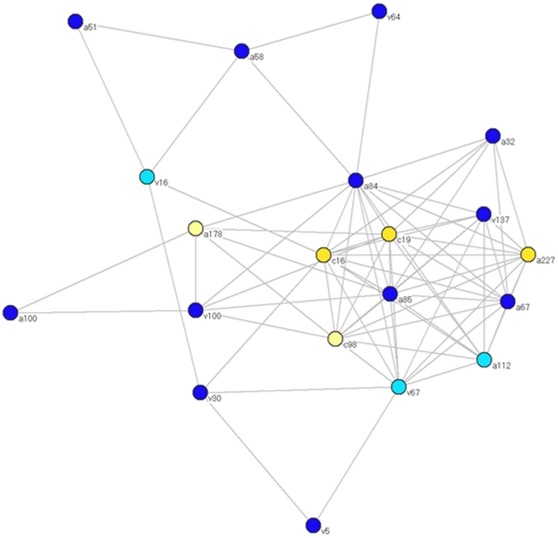
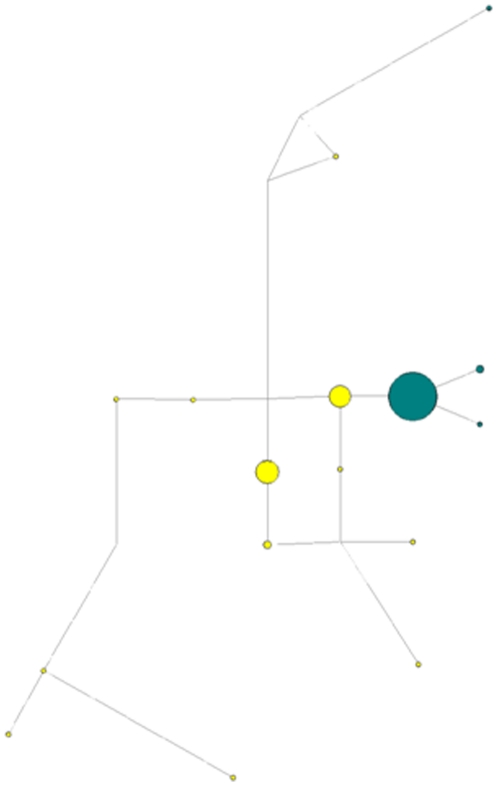
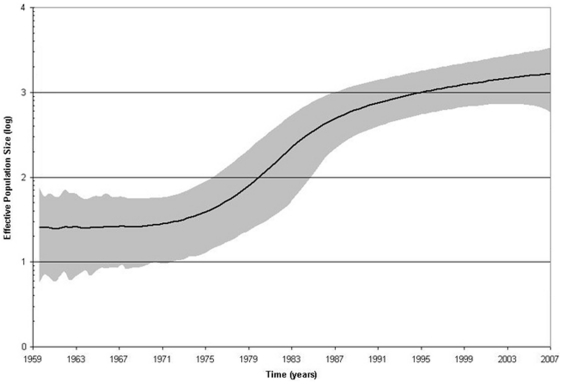
Methods and findings: Despite remoteness and isolation, approximately 11% of the entire population in these communities was HBV-DNA seropositive. Analyses of the S-gene sequences obtained from 55 HBV-seropositive individuals showed the circulation of 37 distinct HBV variants. These HBV isolates belong predominantly to genotype E (HBV/E) (n=53, 96.4%), with only 2 classified as sub-genotype A3 (HBV/A3). Phylogenetic analysis showed extensive intermixing between HBV/E variants identified in these communities and different countries in Africa. Quasispecies analysis of 22 HBV/E strains using end-point limiting-dilution real-time PCR, sequencing and median joining networks showed extensive intra-host heterogeneity and inter-host variant sharing. To investigate events that resulted in such remarkable HBV/E diversity, HBV full-size genome sequences were obtained from 47 HBV/E infected persons and P gene was subjected to Bayesian coalescent analysis. The time to the most recent common ancestor (tMRCA) for these HBV/E variants was estimated to be year 1952 (95% highest posterior density (95% HPD): 1927-1970). Using additional HBV/E sequences from other African countries, the tMRCA was estimated to be year 1948 (95% HPD: 1924-1966), indicating that HBV/E in these remote communities has a similar time of origin with multiple HBV/E variants broadly circulating in West/Central Africa. Phylogenetic analysis and statistical neutrality tests suggested rapid HBV/E population expansion. Additionally, skyline plot analysis showed an increase in the size of the HBV/E-infected population over the last approximately 30-40 years.
Conclusions: Our data suggest a massive introduction and relatively recent HBV/E expansion in the human population in Africa. Collectively, these data show a significant shift in the HBV/E epidemic dynamics in Africa over the last century.
Conflict of interest statement
Figures

References
-
- Goldstein ST, Zhou F, Hadler SC, Bell BP, Mast EE, et al. A mathematical model to estimate global hepatitis B disease burden and vaccination impact. Int J Epidemiol. 2005;34:1329–1339. - PubMed
-
- Lee WM. Hepatitis B virus infection. N Engl J Med. 1997;337:1733–1745. - PubMed
-
- Zanetti AR, Van Damme P, Shouval D. The global impact of vaccination against hepatitis B: a historical overview. Vaccine. 2008;26:6266–6273. - PubMed
-
- Ganem D, Schneider RJ. Hepadnaviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 2923–2969.
-
- Norder H, Courouce AM, Coursaget P, Echevarria JM, Lee SD, et al. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology. 2004;47:289–309. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
