

Liquid-liquid two-phase systems for the production of porous hydrogels and hydrogel microspheres for biomedical applications: A tutorial review
- PMID: 20659596
- PMCID: PMC2967636
- DOI: 10.1016/j.actbio.2010.07.028
Liquid-liquid two-phase systems for the production of porous hydrogels and hydrogel microspheres for biomedical applications: A tutorial review
Abstract
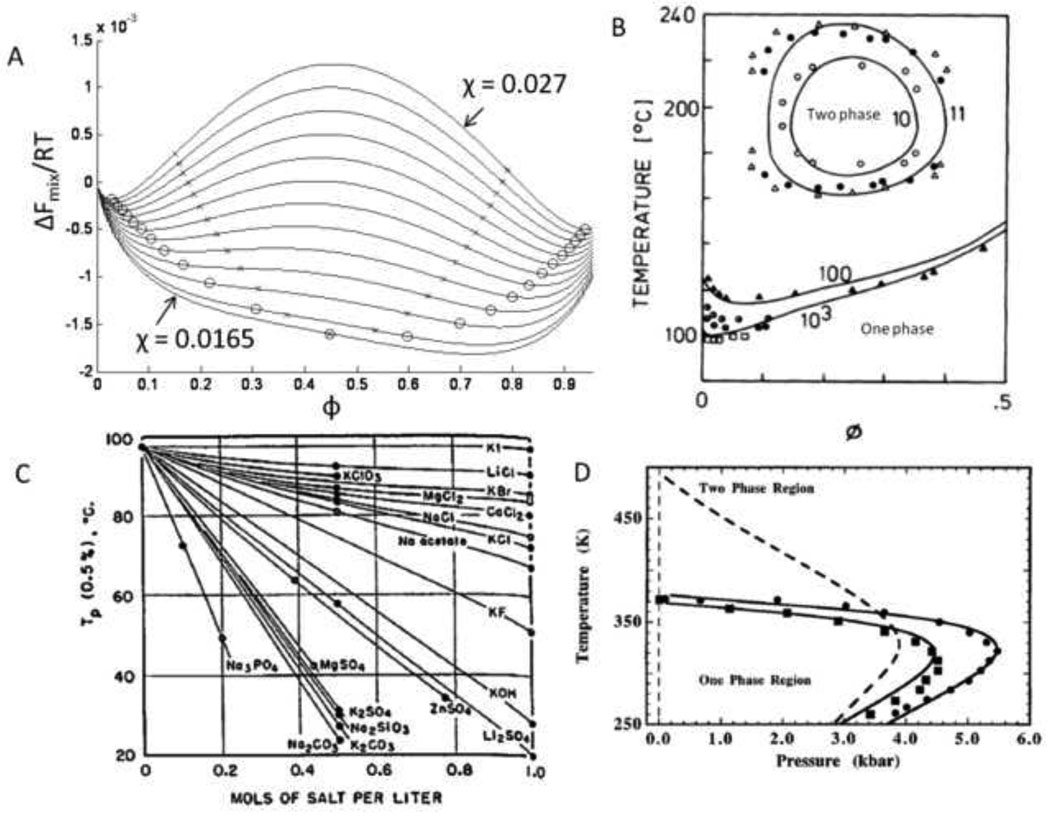
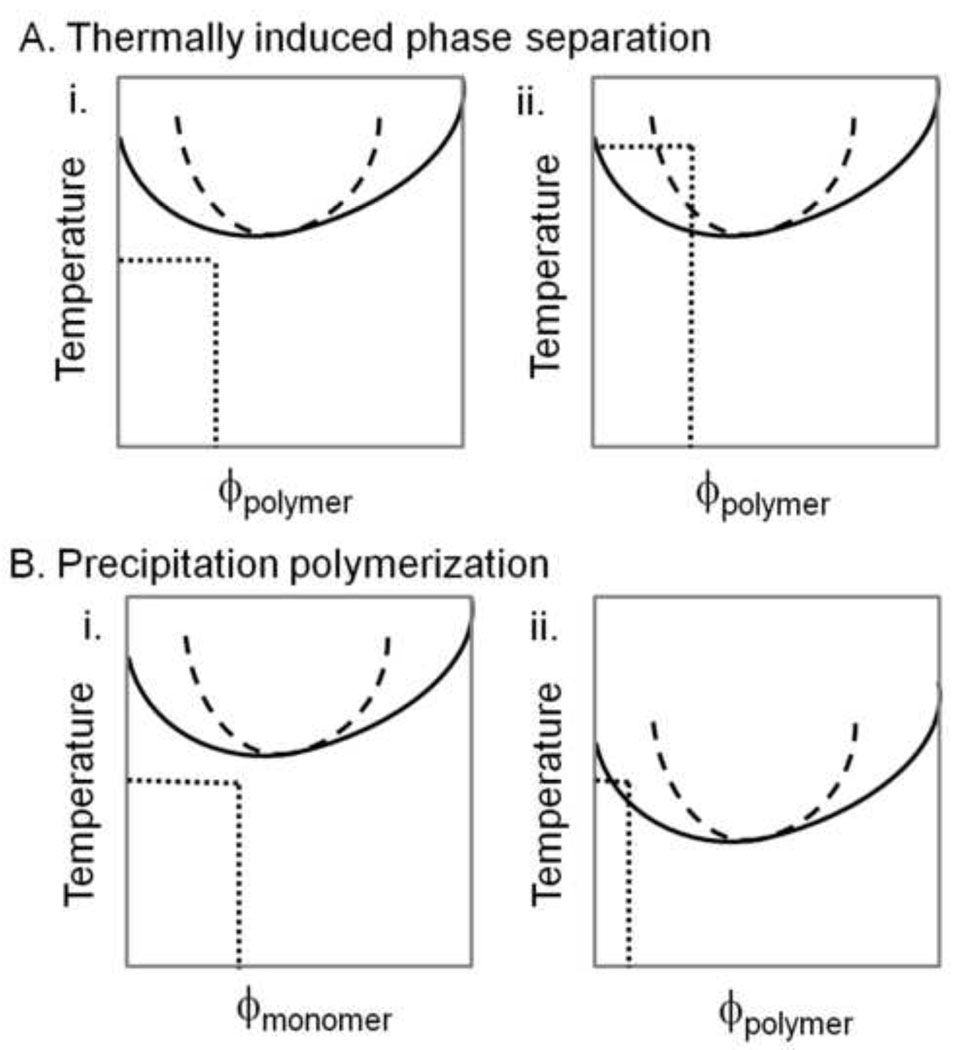
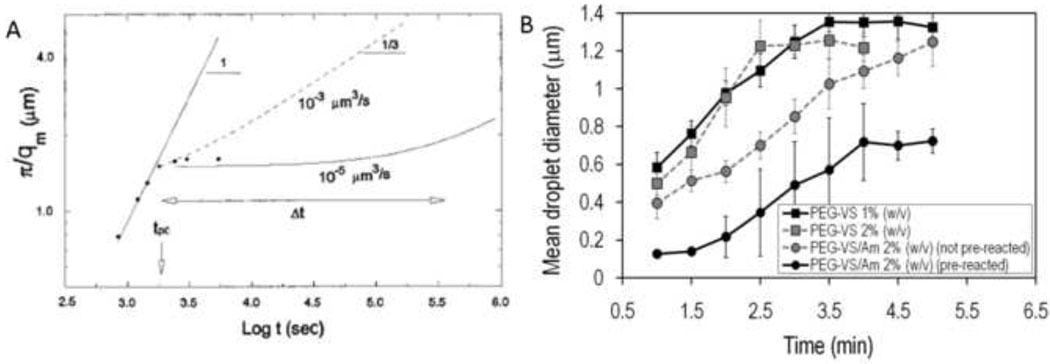
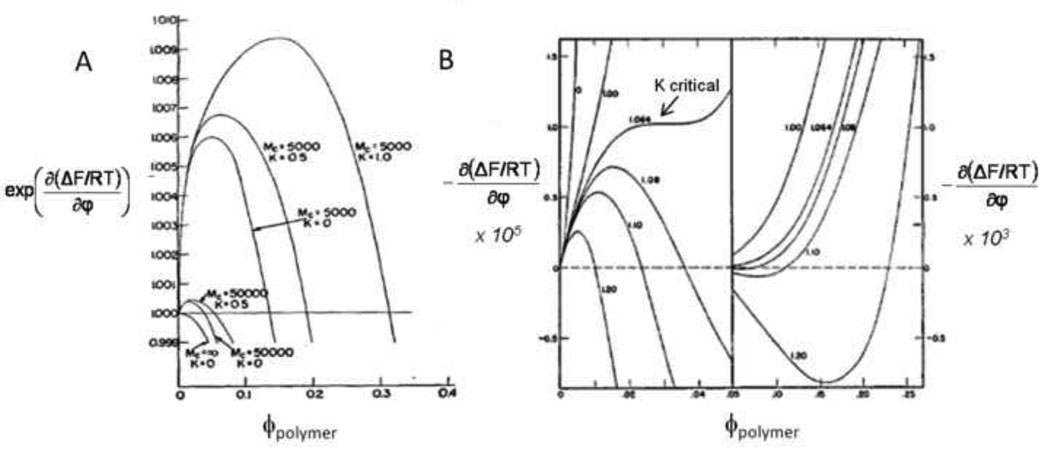
Macroporous hydrogels may have direct applications in regenerative medicine as scaffolds to support tissue formation. Hydrogel microspheres may be used as drug-delivery vehicles or as building blocks to assemble modular scaffolds. A variety of techniques exist to produce macroporous hydrogels and hydrogel microspheres. A subset of these relies on liquid-liquid two-phase systems. Within this subset, vastly different types of polymerization processes are found. In this review, the history, terminology and classification of liquid-liquid two-phase polymerization and crosslinking are described. Instructive examples of hydrogel microsphere and macroporous scaffold formation by precipitation/dispersion, emulsion and suspension polymerizations are used to illustrate the nature of these processes. The role of the kinetics of phase separation in determining the morphology of scaffolds and microspheres is also delineated. Brief descriptions of miniemulsion, microemulsion polymerization and ionotropic gelation are also included.
Copyright © 2010 Acta Materialia Inc. Published by Elsevier Ltd. All rights reserved.
Figures



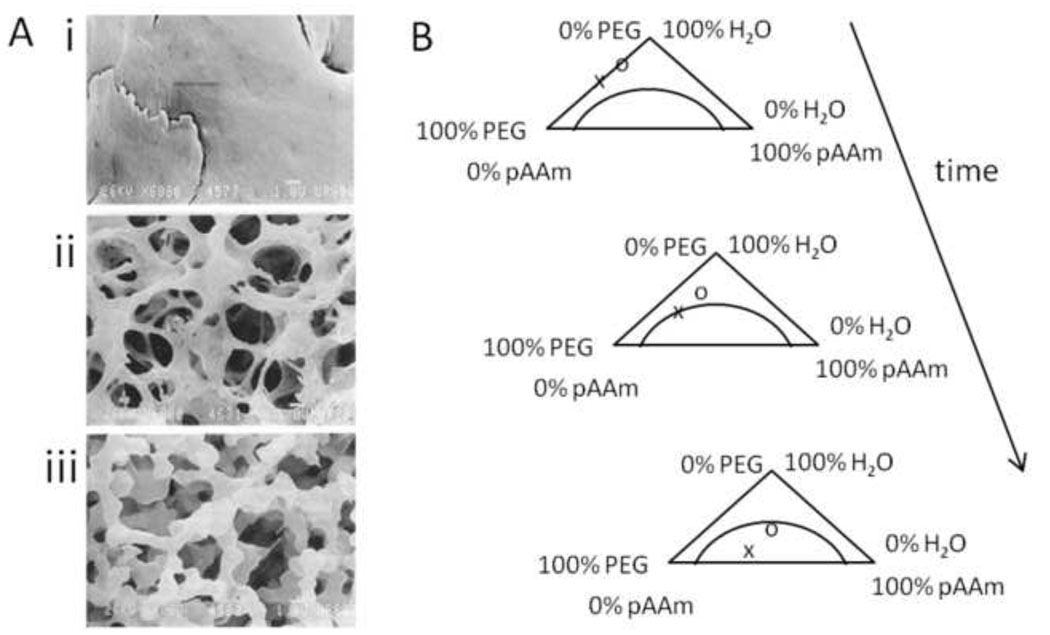



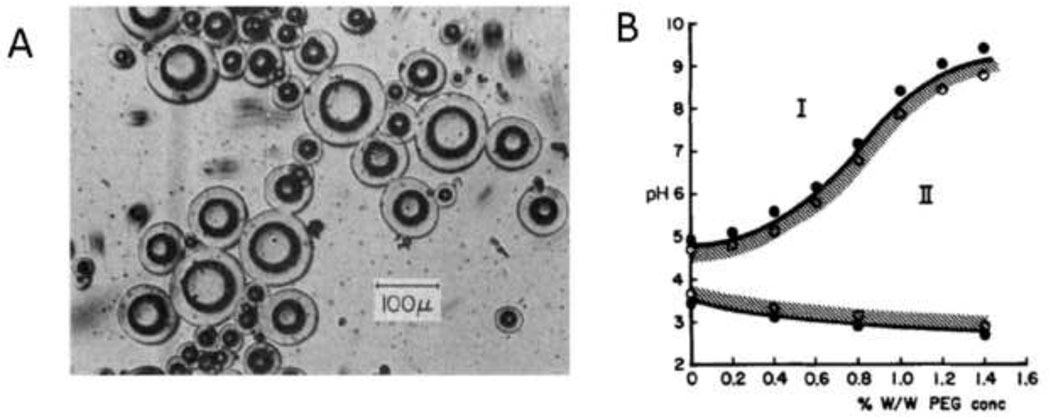

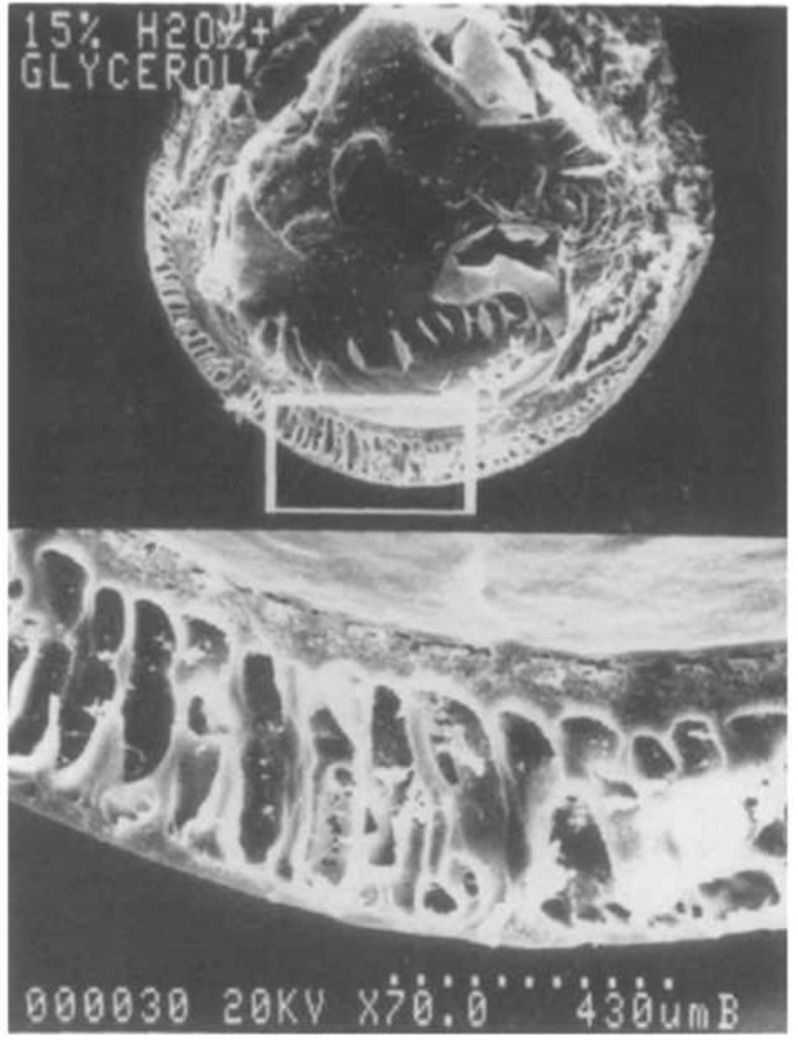
References
-
- Chung HJ, Park TG. Surface engineered and drug releasing pre-fabricated scaffolds for tissue engineering. Advanced Drug Delivery Reviews. 2007;59:249–262. - PubMed
-
- Karageorgiou V, Kaplan D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials. 2005;26:5474–5491. - PubMed
-
- Mikos AG, Temenoff JS. Formation of highly porous biodegradable scaffolds for tissue engineering. EJB Electronic Journal of Biotechnology. 2000;3:114–119.
-
- Rivest C, Morrison DWG, Ni B, Rubin J, Yadav V, Mahdavi A, et al. Microscale hydrogels for medicine and biology: Synthesis, characteristics and applications. Journal of Mechanics of Materials and Structures. 2007;2:1103–1119.
-
- Khademhosseini A, Langer R. Microengineered hydrogels for tissue engineering. Biomaterials. 2007;28:5087–5092. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
