

Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic
- PMID: 20660316
- PMCID: PMC2922607
- DOI: 10.1073/pnas.1002713107
Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic
Abstract
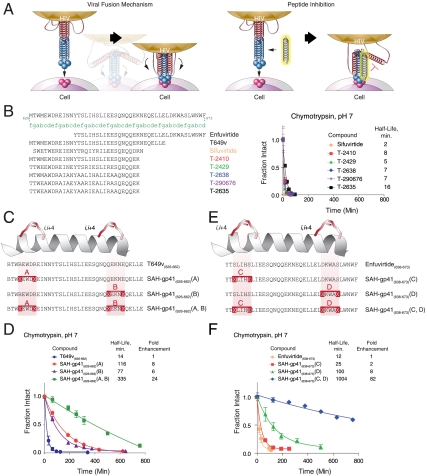
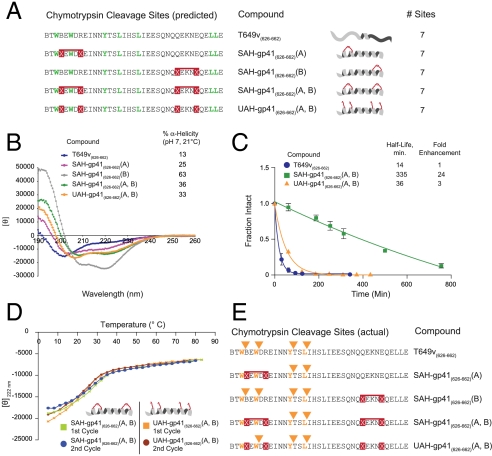
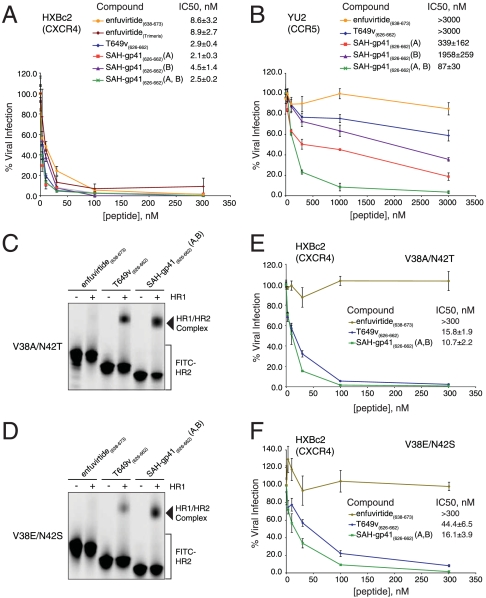
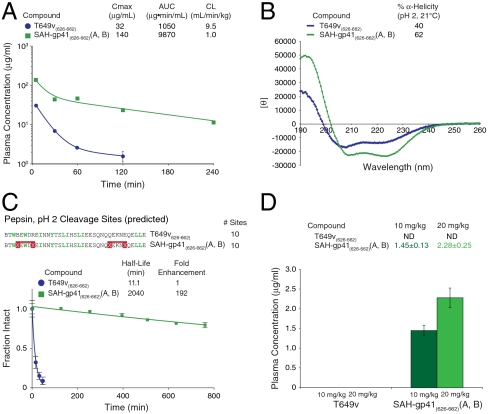
The pharmacologic utility of lengthy peptides can be hindered by loss of bioactive structure and rapid proteolysis, which limits bioavailability. For example, enfuvirtide (Fuzeon, T20, DP178), a 36-amino acid peptide that inhibits human immunodeficiency virus type 1 (HIV-1) infection by effectively targeting the viral fusion apparatus, has been relegated to a salvage treatment option mostly due to poor in vivo stability and lack of oral bioavailability. To overcome the proteolytic shortcomings of long peptides as therapeutics, we examined the biophysical, biological, and pharmacologic impact of inserting all-hydrocarbon staples into an HIV-1 fusion inhibitor. We find that peptide double-stapling confers striking protease resistance that translates into markedly improved pharmacokinetic properties, including oral absorption. We determined that the hydrocarbon staples create a proteolytic shield by combining reinforcement of overall alpha-helical structure, which slows the kinetics of proteolysis, with complete blockade of peptide cleavage at constrained sites in the immediate vicinity of the staple. Importantly, double-stapling also optimizes the antiviral activity of HIV-1 fusion peptides and the antiproteolytic feature extends to other therapeutic peptide templates, such as the diabetes drug exenatide (Byetta). Thus, hydrocarbon double-stapling may unlock the therapeutic potential of natural bioactive polypeptides by transforming them into structurally fortified agents with enhanced bioavailability.
Conflict of interest statement
Conflict of interest statement: L.D.W. is a consultant and scientific advisory board member for Aileron Therapeutics.
Figures
References
-
- Blackwell HE, Grubbs RH. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew Chem Int Edit. 1998;37:3281–3284. - PubMed
-
- Blackwell HE, et al. Ring-closing metathesis of olefinic peptides: Design, synthesis, and structural characterization of macrocyclic helical peptides. J Org Chem. 2001;66:5291–5302. - PubMed
-
- Schafmeister CE, Po J, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J Am Chem Soc. 2000;122:5891–5892.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
