

Polycyclic aromatic hydrocarbon components contribute to the mitochondria-antiapoptotic effect of fine particulate matter on human bronchial epithelial cells via the aryl hydrocarbon receptor
- PMID: 20663163
- PMCID: PMC2914693
- DOI: 10.1186/1743-8977-7-18
Polycyclic aromatic hydrocarbon components contribute to the mitochondria-antiapoptotic effect of fine particulate matter on human bronchial epithelial cells via the aryl hydrocarbon receptor
Abstract
Background: Nowadays, effects of fine particulate matter (PM2.5) are well-documented and related to oxidative stress and pro-inflammatory response. Nevertheless, epidemiological studies show that PM2.5 exposure is correlated with an increase of pulmonary cancers and the remodeling of the airway epithelium involving the regulation of cell death processes. Here, we investigated the components of Parisian PM2.5 involved in either the induction or the inhibition of cell death quantified by different parameters of apoptosis and delineated the mechanism underlying this effect.
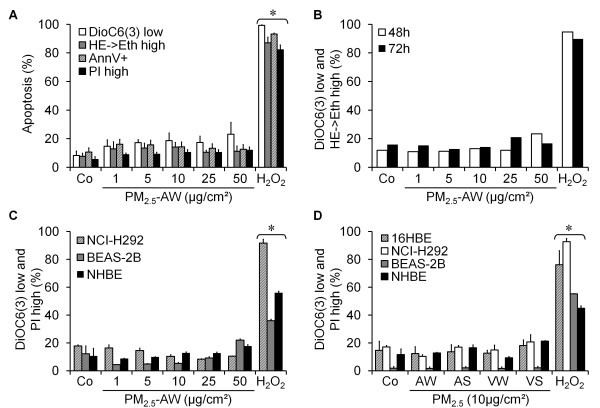
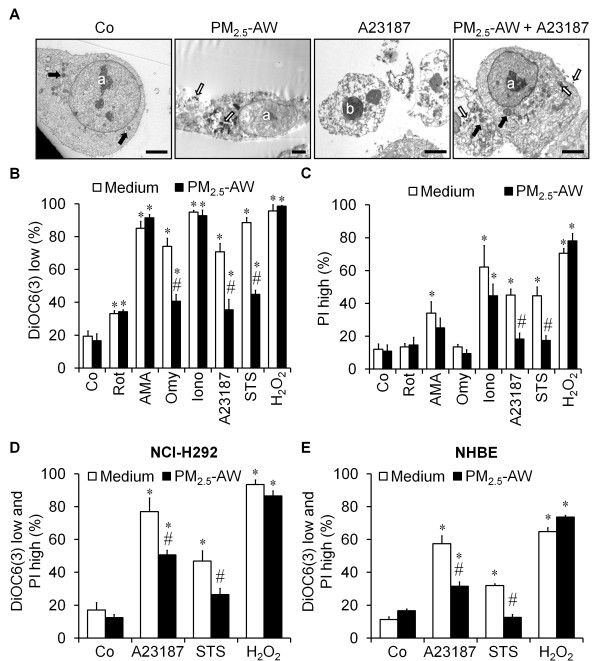
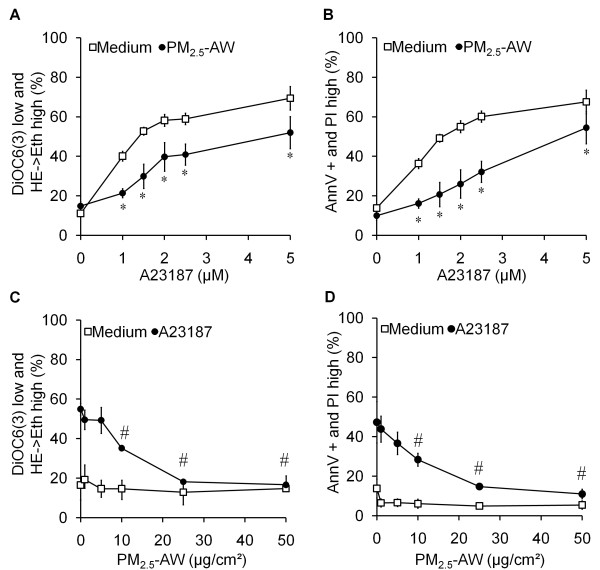
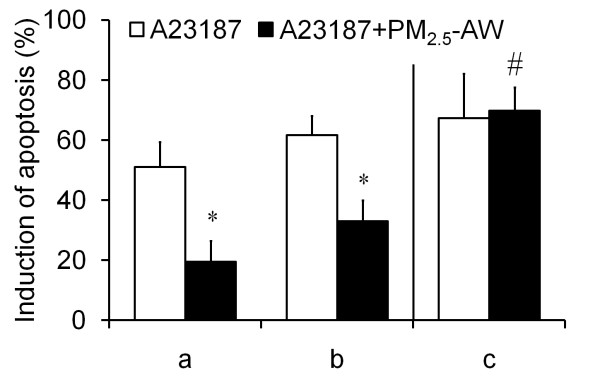
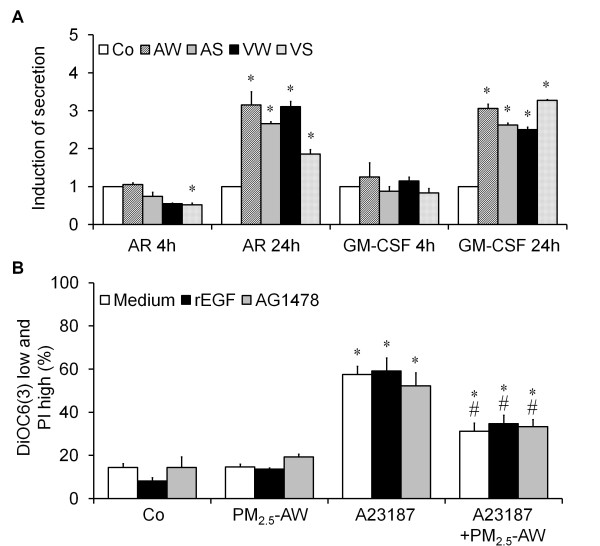
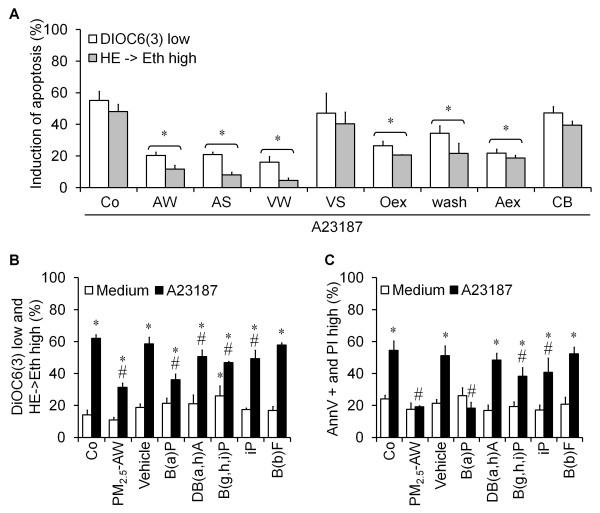
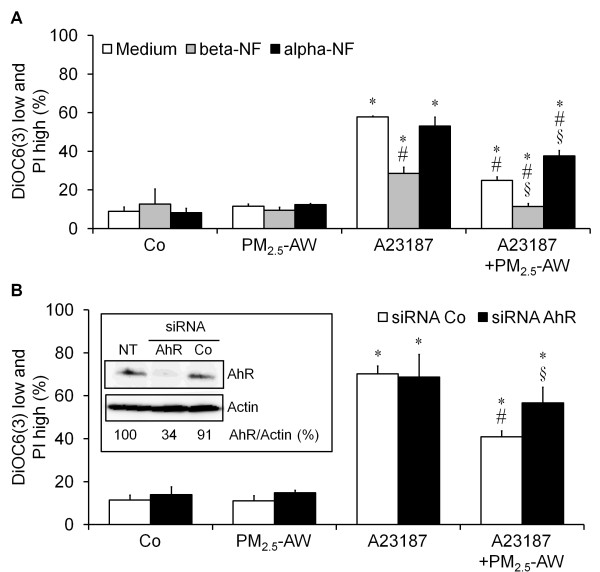
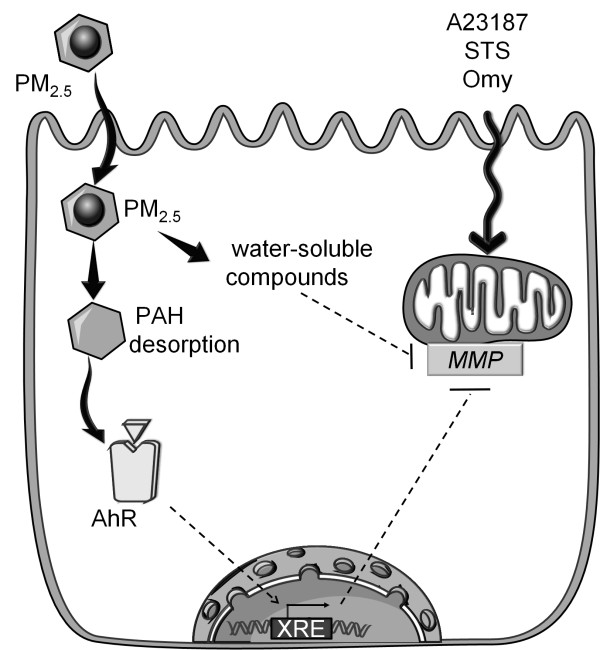
Results: In this study, we showed that low levels of Parisian PM2.5 are not cytotoxic for three different cell lines and primary cultures of human bronchial epithelial cells. Conversely, a 4 hour-pretreatment with PM2.5 prevent mitochondria-driven apoptosis triggered by broad spectrum inducers (A23187, staurosporine and oligomycin) by reducing the mitochondrial transmembrane potential loss, the subsequent ROS production, phosphatidylserine externalization, plasma membrane permeabilization and typical morphological outcomes (cell size decrease, massive chromatin and nuclear condensation, formation of apoptotic bodies). The use of recombinant EGF and specific inhibitor led us to rule out the involvement of the classical EGFR signaling pathway as well as the proinflammatory cytokines secretion. Experiments performed with different compounds of PM2.5 suggest that endotoxins as well as carbon black do not participate to the antiapoptotic effect of PM2.5. Instead, the water-soluble fraction, washed particles and organic compounds such as polycyclic aromatic hydrocarbons (PAH) could mimic this antiapoptotic activity. Finally, the activation or silencing of the aryl hydrocarbon receptor (AhR) showed that it is involved into the molecular mechanism of the antiapoptotic effect of PM2.5 at the mitochondrial checkpoint of apoptosis.
Conclusions: The PM2.5-antiapoptotic effect in addition to the well-documented inflammatory response might explain the maintenance of a prolonged inflammation state induced after pollution exposure and might delay repair processes of injured tissues.
Figures
Similar articles
-
Lung cancer associated with combustion particles and fine particulate matter (PM2.5) - The roles of polycyclic aromatic hydrocarbons (PAHs) and the aryl hydrocarbon receptor (AhR).Biochem Pharmacol. 2023 Oct;216:115801. doi: 10.1016/j.bcp.2023.115801. Epub 2023 Sep 9. Biochem Pharmacol. 2023. PMID: 37696458 Free PMC article. Review.
-
Pro-inflammatory response and oxidative stress induced by specific components in ambient particulate matter in human bronchial epithelial cells.Environ Toxicol. 2016 Aug;31(8):923-36. doi: 10.1002/tox.22102. Epub 2014 Dec 23. Environ Toxicol. 2016. PMID: 25533354
-
Fine particulate matter induces amphiregulin secretion by bronchial epithelial cells.Am J Respir Cell Mol Biol. 2004 Apr;30(4):421-7. doi: 10.1165/rcmb.2003-0281RC. Epub 2003 Dec 30. Am J Respir Cell Mol Biol. 2004. PMID: 14701705
-
TNFα and IL-6 Responses to Particulate Matter in Vitro: Variation According to PM Size, Season, and Polycyclic Aromatic Hydrocarbon and Soil Content.Environ Health Perspect. 2016 Apr;124(4):406-12. doi: 10.1289/ehp.1409287. Epub 2015 Sep 15. Environ Health Perspect. 2016. PMID: 26372663 Free PMC article.
-
Potential role of PM2.5 in melanogenesis.Environ Int. 2019 Nov;132:105063. doi: 10.1016/j.envint.2019.105063. Epub 2019 Aug 6. Environ Int. 2019. PMID: 31394397 Review.
Cited by
-
The Road to Malignant Cell Transformation after Particulate Matter Exposure: From Oxidative Stress to Genotoxicity.Int J Mol Sci. 2023 Jan 16;24(2):1782. doi: 10.3390/ijms24021782. Int J Mol Sci. 2023. PMID: 36675297 Free PMC article. Review.
-
Functional Effects of Cigarette Smoke-Induced Changes in Airway Smooth Muscle Mitochondrial Morphology.J Cell Physiol. 2017 May;232(5):1053-1068. doi: 10.1002/jcp.25508. Epub 2016 Sep 21. J Cell Physiol. 2017. PMID: 27474898 Free PMC article.
-
Associations between Urinary Concentrations of Polycyclic Aromatic Hydrocarbons and Overactive Bladder in US Adults: Data from the National Health and Nutrition Examination Survey 2005-2016.Urol Int. 2024;108(2):137-145. doi: 10.1159/000536253. Epub 2024 Jan 12. Urol Int. 2024. PMID: 38219726 Free PMC article.
-
Lung cancer associated with combustion particles and fine particulate matter (PM2.5) - The roles of polycyclic aromatic hydrocarbons (PAHs) and the aryl hydrocarbon receptor (AhR).Biochem Pharmacol. 2023 Oct;216:115801. doi: 10.1016/j.bcp.2023.115801. Epub 2023 Sep 9. Biochem Pharmacol. 2023. PMID: 37696458 Free PMC article. Review.
-
Deciphering the Code between Air Pollution and Disease: The Effect of Particulate Matter on Cancer Hallmarks.Int J Mol Sci. 2019 Dec 24;21(1):136. doi: 10.3390/ijms21010136. Int J Mol Sci. 2019. PMID: 31878205 Free PMC article. Review.
References
-
- Boland S, Baeza-Squiban A, Marano F. Toxicité respiratoire des particules Diesel: les mécanismes cellulaires et moléculaires. Médecine Sciences. 2001;17:596–603.
-
- Baulig A, Poirault JJ, Ausset P, Schins R, Shi T, Baralle D, Dorlhene P, Meyer M, Lefevre R, Baeza-Squiban A, Marano F. Physicochemical characteristics and biological activities of seasonal atmospheric particulate matter sampling in two locations of Paris. Environ Sci Technol. 2004;38:5985–5992. doi: 10.1021/es049476z. - DOI - PubMed
-
- Baulig A, Garlatti M, Bonvallot V, Marchand A, Barouki R, Marano F, Baeza-Squiban A. Involvement of reactive oxygen species in the metabolic pathways triggered by diesel exhaust particles in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L671–679. - PubMed
-
- Boland S, Baeza-Squiban A, Fournier T, Houcine O, Gendron MC, Chevrier M, Jouvenot G, Coste A, Aubier M, Marano F. Diesel exhaust particles are taken up by human airway epithelial cells in vitro and alter cytokine production. Am J Physiol. 1999;276:L604–613. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
