

The proliferating role of insulin and insulin-like growth factors in cancer
- PMID: 20663687
- PMCID: PMC2949481
- DOI: 10.1016/j.tem.2010.06.007
The proliferating role of insulin and insulin-like growth factors in cancer
Abstract
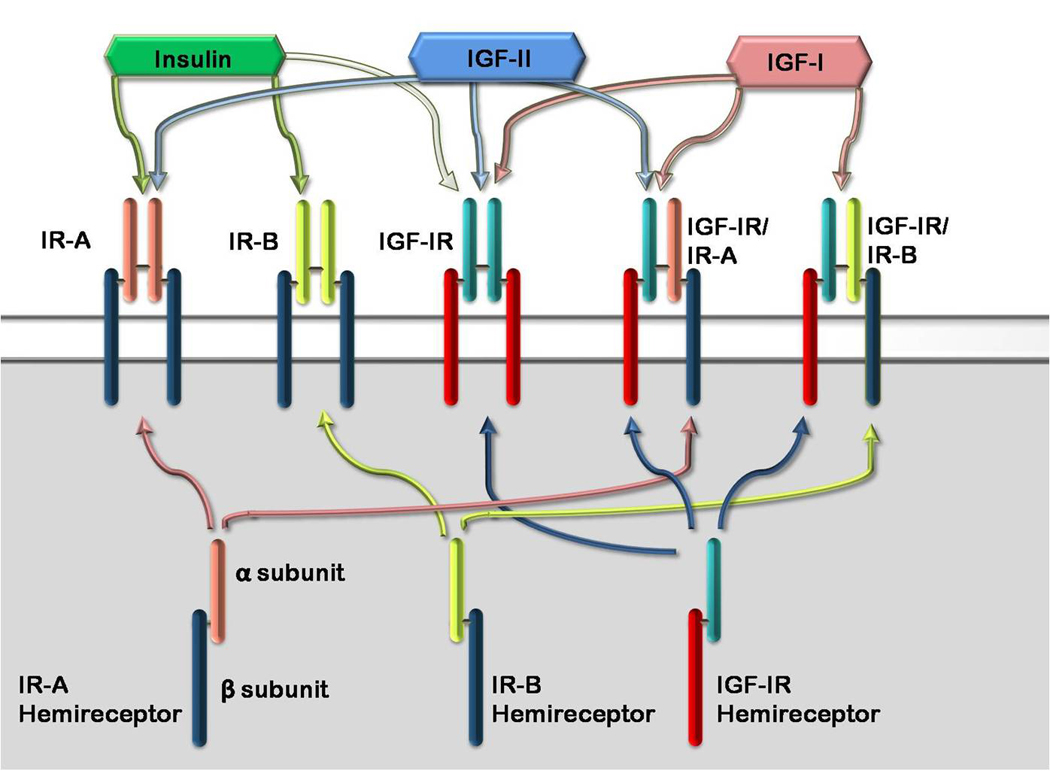
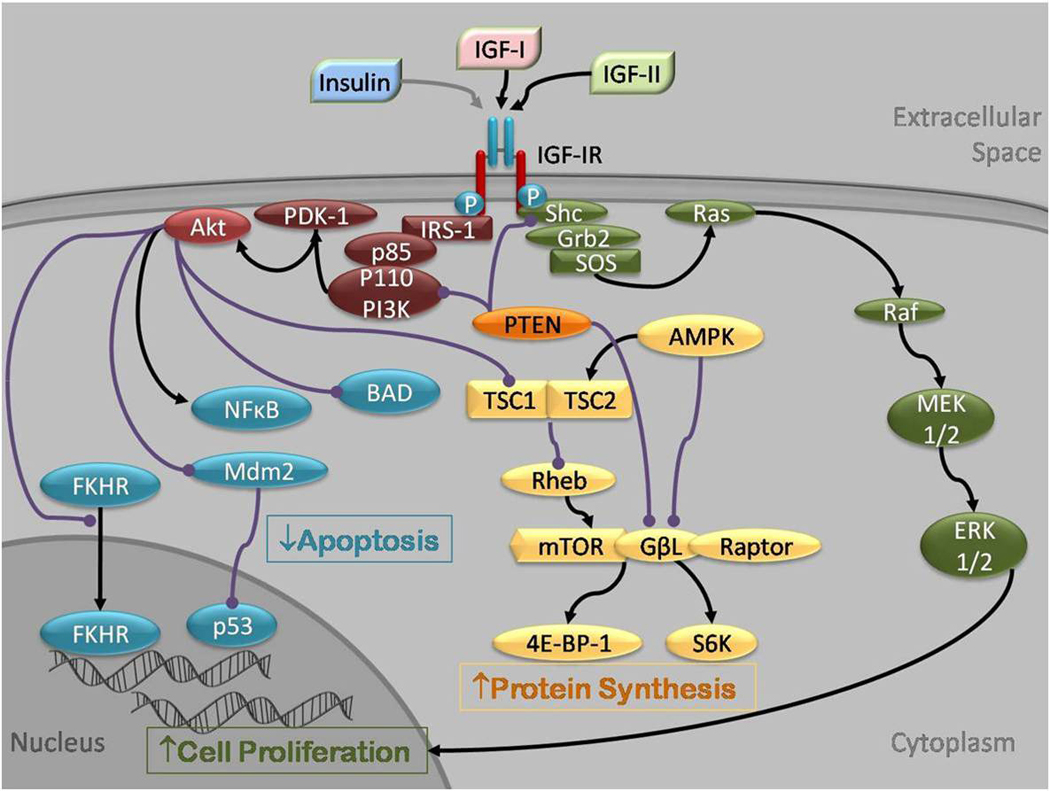
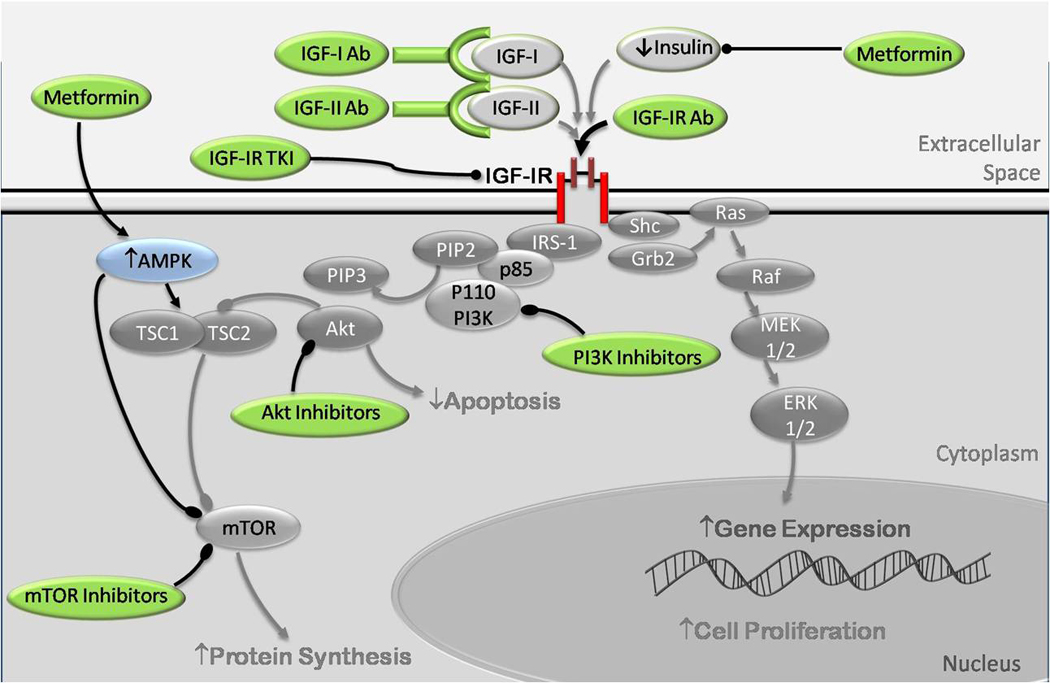
Epidemiological studies have reported an increased risk of cancer in people with type 2 diabetes (T2DM) and obesity, related in part to hyperinsulinemia, secondary to insulin resistance. Hyperinsulinemia leads to increased expression of insulin-like growth factor (IGF)-I expression. In fact, increased insulin, IGF-I and IGF-II levels are associated with tumor growth in vitro, in animal models, and in epidemiological studies in humans. In this paper, we discuss the roles of insulin, IGF-I and IGF-II, their interaction with the insulin receptor (IR) and IGF-I receptor (IGF-IR), and their signaling pathways and regulation as these pertain to tumor growth. We explain how these pathways have been deciphered by in vitro and in vivo studies, and how they are being exploited in the development of targeted cancer therapies.
Copyright © 2010 Elsevier Ltd. All rights reserved.
Figures
References
-
- Vigneri P, et al. Diabetes and cancer. Endocr Relat Cancer. 2009;16(4):1103–1123. - PubMed
-
- Calle EE, et al. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N Engl J Med. 2003;348(17):1625–1638. - PubMed
-
- Flegal KM, et al. Prevalence and Trends in Obesity Among US Adults, 1999–2008. JAMA. 2010;303(3):235–241. - PubMed
-
- Currie C, Poole C, Gale E. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52(9):1766–1777. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
