

Development of a novel tumor-targeted vascular disrupting agent activated by membrane-type matrix metalloproteinases
- PMID: 20663911
- PMCID: PMC2933508
- DOI: 10.1158/0008-5472.CAN-10-1440
Development of a novel tumor-targeted vascular disrupting agent activated by membrane-type matrix metalloproteinases
Abstract
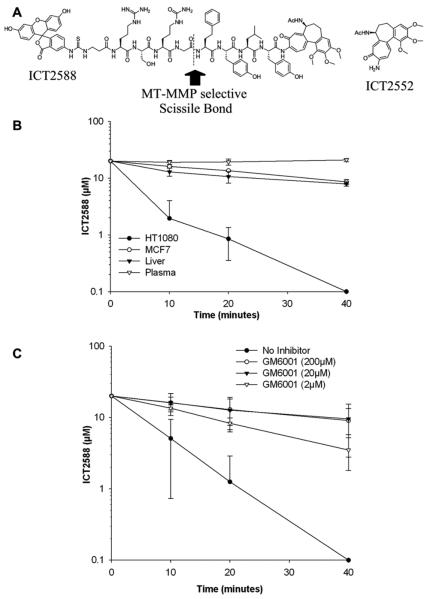
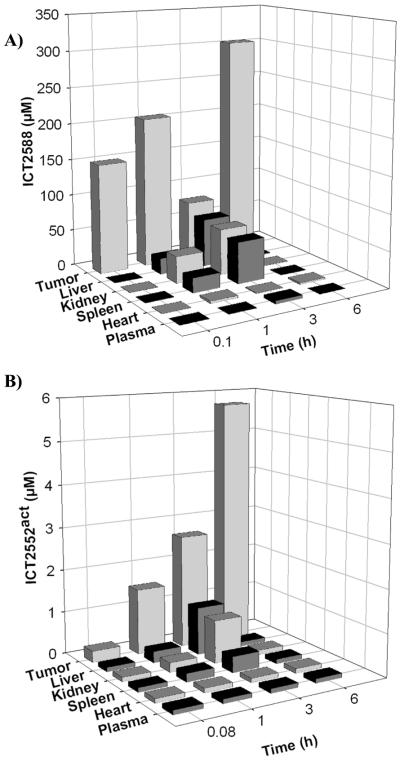
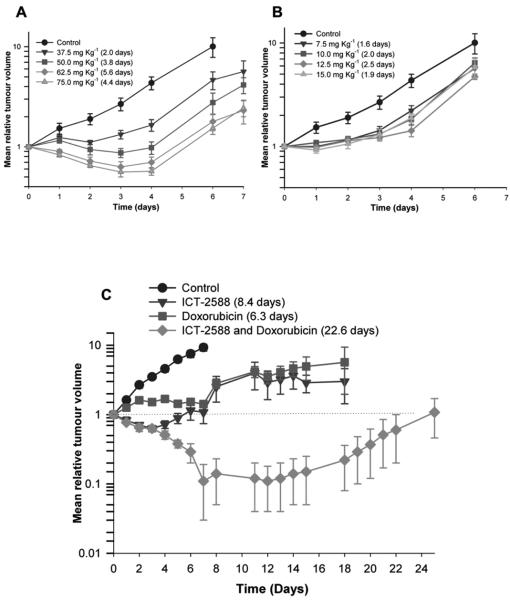
Vascular disrupting agents (VDA) offer a strategy to starve solid tumors of nutrients and oxygen concomitant with tumor shrinkage. Several VDAs have progressed into early clinical trials, but their therapeutic value seems to be compromised by systemic toxicity. In this report, we describe the design and characterization of a novel VDA, ICT2588, that is nontoxic until activated specifically in the tumor by membrane-type 1 matrix metalloproteinase (MT1-MMP). HT1080 cancer cells expressing MT1-MMP were selectively chemosensitive to ICT2588, whereas MCF7 cells that did not express MT1-MMP were nonresponsive. Preferential hydrolysis of ICT2588 to its active metabolite (ICT2552) was observed in tumor homogenates of HT1080 relative to MCF7 homogenates, mouse plasma, and liver homogenate. ICT2588 activation was inhibited by the MMP inhibitor ilomastat. In HT1080 tumor-bearing mice, ICT2588 administration resulted in the formation of the active metabolite, diminution of tumor vasculature, and hemorrhagic necrosis of the tumor. The antitumor activity of ICT2588 was superior to its active metabolite, exhibiting reduced toxicity, improved therapeutic index, enhanced pharmacodynamic effect, and greater efficacy. Coadministration of ICT2588 with doxorubicin resulted in a significant antitumor response (22.6 d growth delay), which was superior to the administration of ICT2588 or doxorubicin as a single agent, including complete tumor regressions. Our findings support the clinical development of ICT2588, which achieves selective VDA targeting based on MT-MMP activation in the tumor microenvironment.
Figures


Similar articles
-
Tumor-targeted prodrug ICT2588 demonstrates therapeutic activity against solid tumors and reduced potential for cardiovascular toxicity.Mol Pharm. 2014 Apr 7;11(4):1294-300. doi: 10.1021/mp400760b. Epub 2014 Mar 27. Mol Pharm. 2014. PMID: 24641451
-
Targeted delivery of a colchicine analogue provides synergy with ATR inhibition in cancer cells.Biochem Pharmacol. 2022 Jul;201:115095. doi: 10.1016/j.bcp.2022.115095. Epub 2022 May 20. Biochem Pharmacol. 2022. PMID: 35598808
-
Matrix metalloproteinase-activated doxorubicin prodrugs inhibit HT1080 xenograft growth better than doxorubicin with less toxicity.Mol Cancer Ther. 2005 May;4(5):751-60. doi: 10.1158/1535-7163.MCT-05-0006. Mol Cancer Ther. 2005. PMID: 15897239
-
Matrix metalloproteinases: new routes to the use of MT1-MMP as a therapeutic target in angiogenesis-related disease.Curr Pharm Des. 2007;13(17):1787-802. doi: 10.2174/138161207780831284. Curr Pharm Des. 2007. PMID: 17584108 Review.
-
MT1-MMP: universal or particular player in angiogenesis?Cancer Metastasis Rev. 2006 Mar;25(1):77-86. doi: 10.1007/s10555-006-7891-z. Cancer Metastasis Rev. 2006. PMID: 16680574 Review.
Cited by
-
Rethinking Brain Cancer Therapy: Tumor Enzyme Activatable Theranostic Nanoparticles.Mol Imaging. 2017 Jan-Dec;16:1536012117730950. doi: 10.1177/1536012117730950. Mol Imaging. 2017. PMID: 28929923 Free PMC article.
-
Comparison of 5-aminolevulinic acid and MMP-14 targeted peptide probes in preclinical models of GBM.Theranostics. 2025 Feb 24;15(8):3517-3531. doi: 10.7150/thno.107210. eCollection 2025. Theranostics. 2025. PMID: 40093889 Free PMC article.
-
A Novel Theranostic Strategy for MMP-14-Expressing Glioblastomas Impacts Survival.Mol Cancer Ther. 2017 Sep;16(9):1909-1921. doi: 10.1158/1535-7163.MCT-17-0022. Epub 2017 Jun 28. Mol Cancer Ther. 2017. PMID: 28659432 Free PMC article.
-
Miniaturized antibodies for imaging membrane type-1 matrix metalloproteinase in cancers.Cancer Sci. 2013 Apr;104(4):495-501. doi: 10.1111/cas.12102. Epub 2013 Feb 17. Cancer Sci. 2013. PMID: 23305265 Free PMC article.
-
The use of thermographic imaging to evaluate therapeutic response in human tumour xenograft models.Sci Rep. 2016 Aug 5;6:31136. doi: 10.1038/srep31136. Sci Rep. 2016. PMID: 27491535 Free PMC article.
References
-
- Lippert JW., 3rd Vascular disrupting agents. Bioorg Med Chem. 2007;15:605–15. - PubMed
-
- Davis PD, Dougherty GJ, Blakey DC, et al. ZD6126: a novel vascular-targeting agent that causes selective destruction of tumor vasculature. Cancer Res. 2002;62:7247–53. - PubMed
-
- Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. 2005;5:423–35. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
