

Role of innate immunity in Helicobacter pylori-induced gastric malignancy
- PMID: 20664074
- PMCID: PMC2990353
- DOI: 10.1152/physrev.00039.2009
Role of innate immunity in Helicobacter pylori-induced gastric malignancy
Abstract
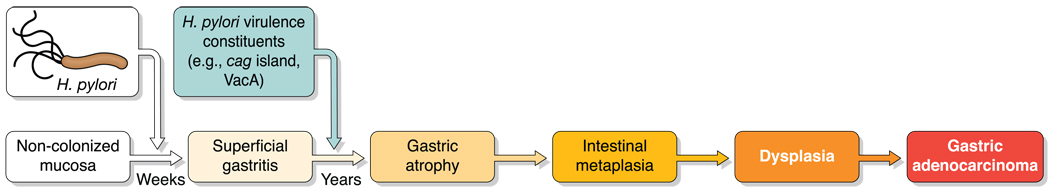
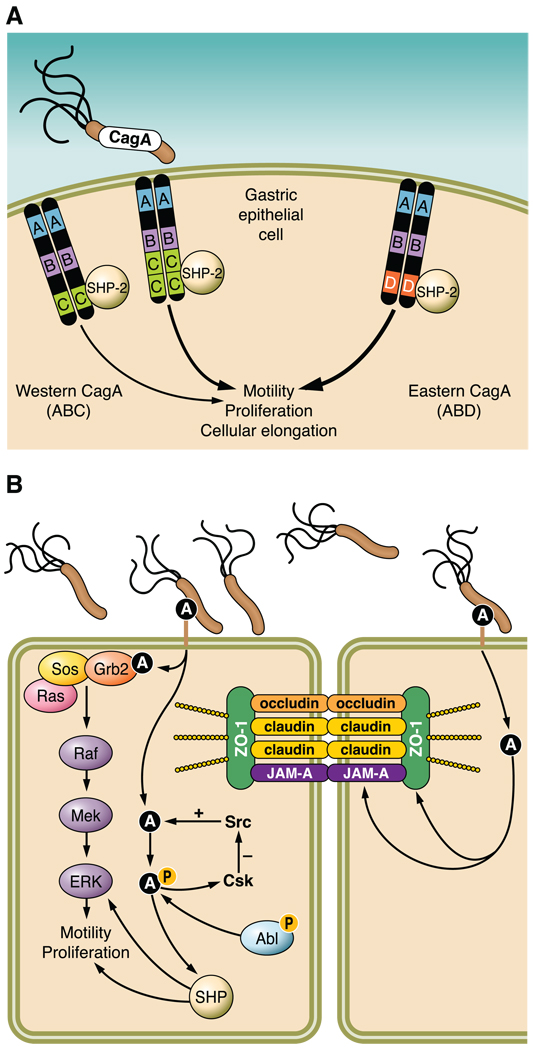
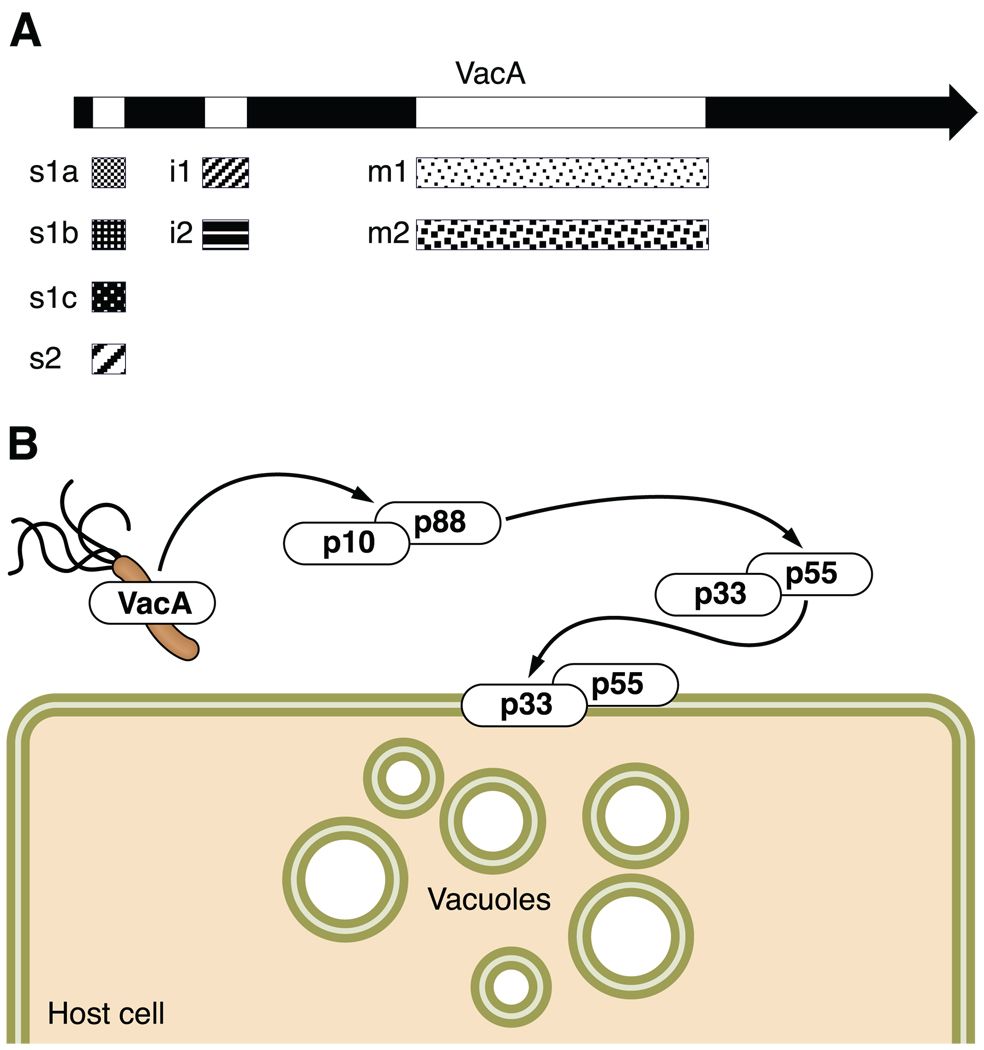
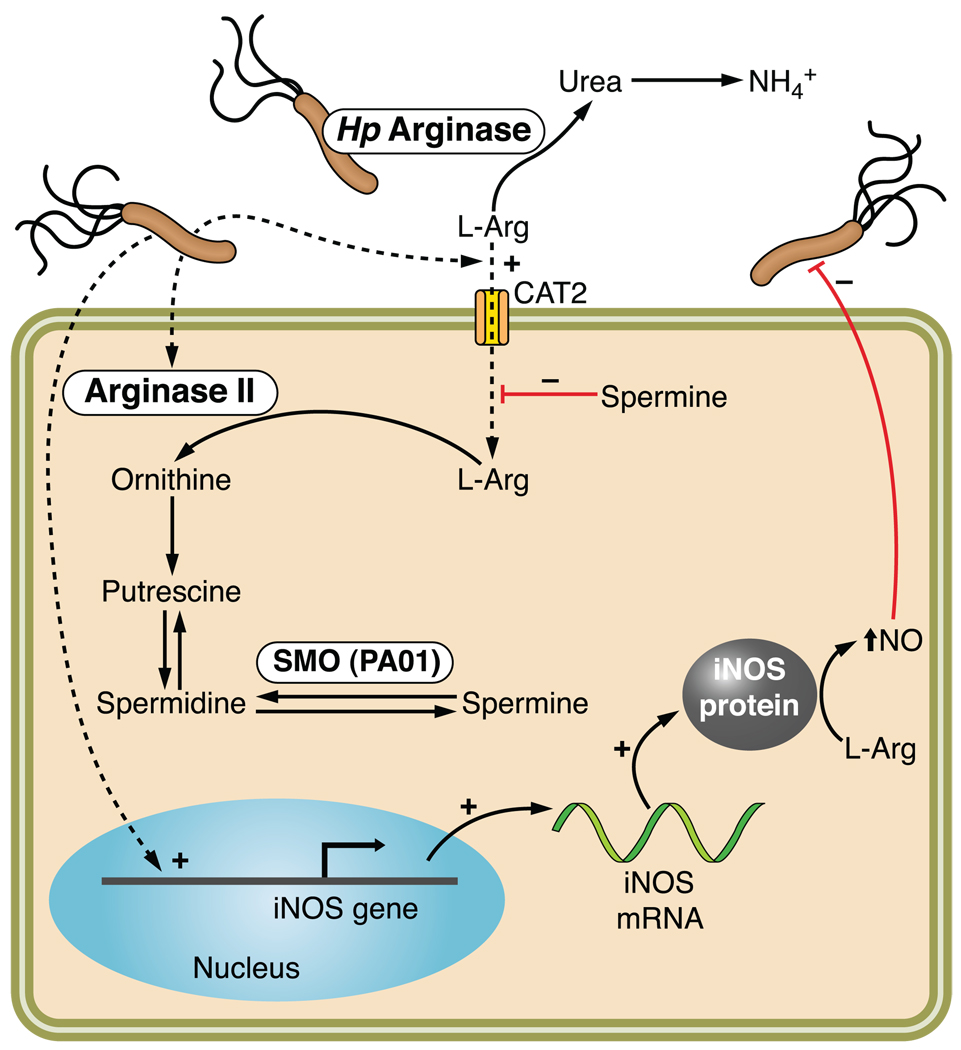
Helicobacter pylori colonizes the majority of persons worldwide, and the ensuing gastric inflammatory response is the strongest singular risk factor for peptic ulceration and gastric cancer. However, only a fraction of colonized individuals ever develop clinically significant outcomes. Disease risk is combinatorial and can be modified by bacterial factors, host responses, and/or specific interactions between host and microbe. Several H. pylori constituents that are required for colonization or virulence have been identified, and their ability to manipulate the host innate immune response will be the focus of this review. Identification of bacterial and host mediators that augment disease risk has profound ramifications for both biomedical researchers and clinicians as such findings will not only provide mechanistic insights into inflammatory carcinogenesis but may also serve to identify high-risk populations of H. pylori-infected individuals who can then be targeted for therapeutic intervention.
Figures


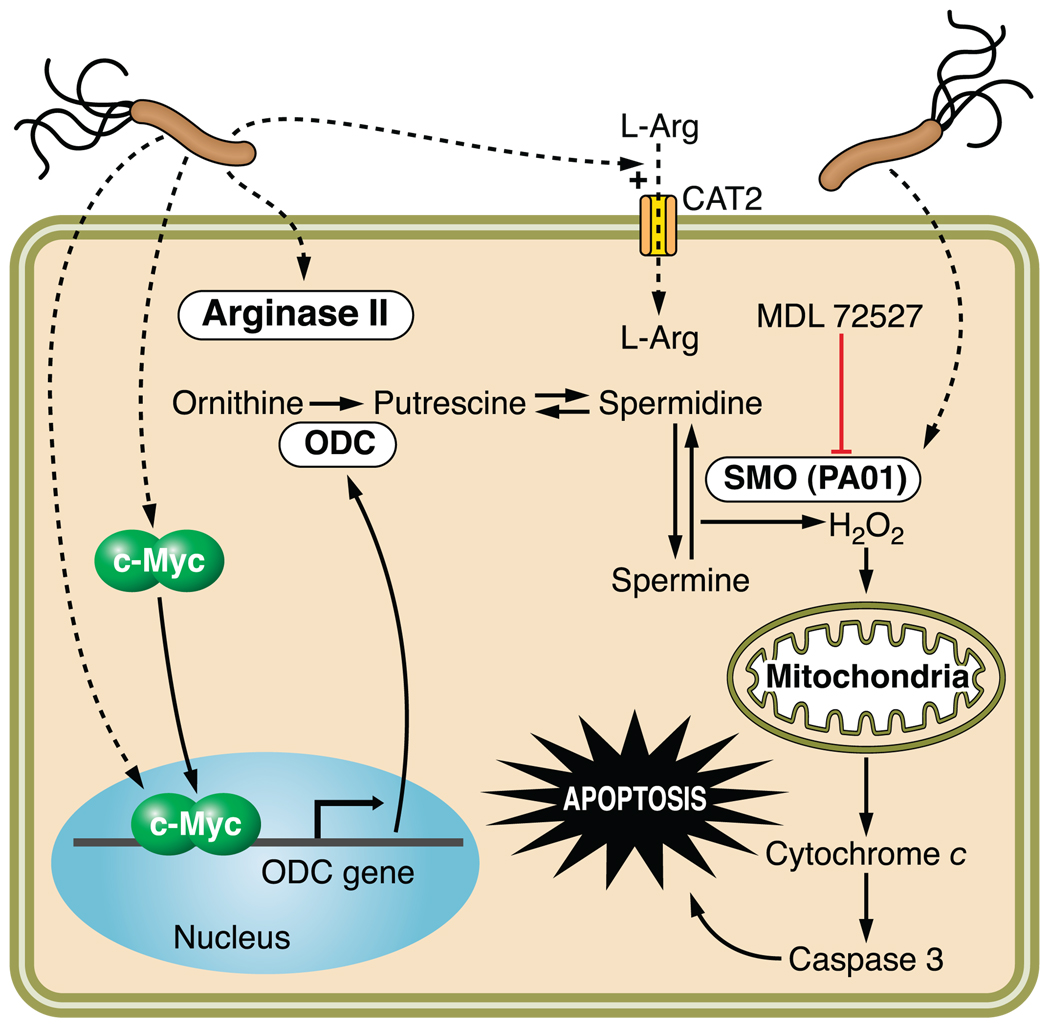
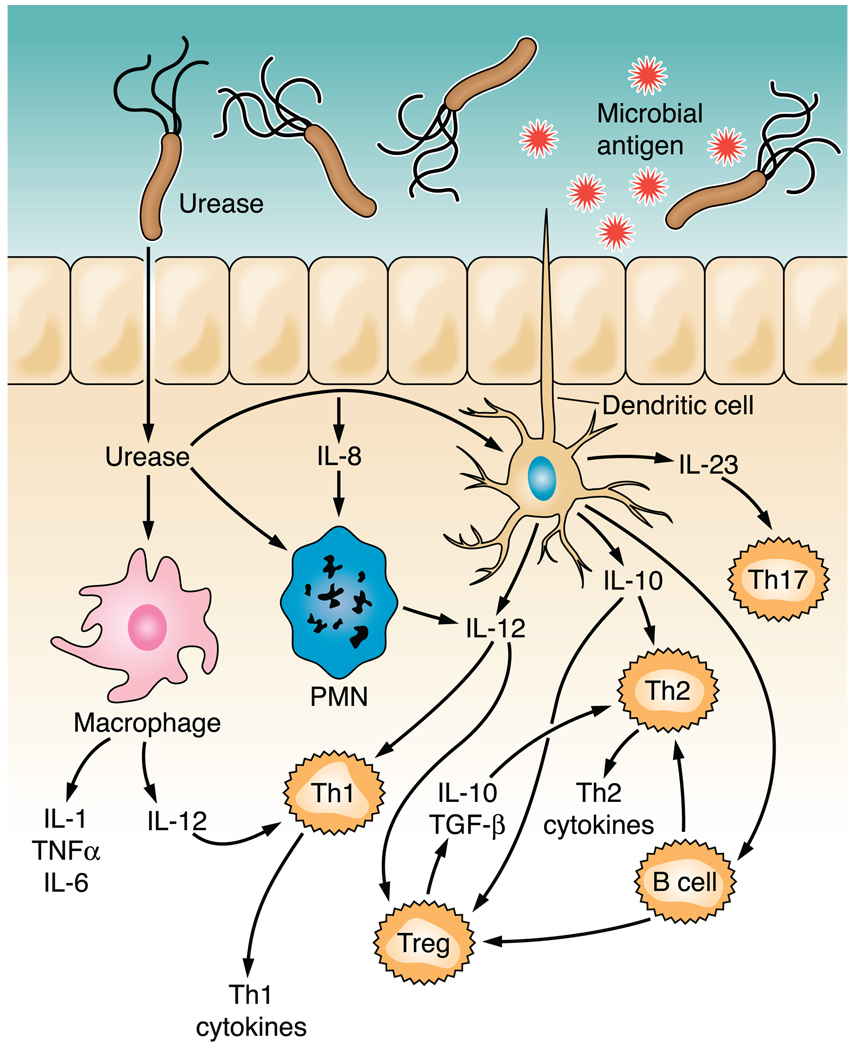
References
-
- Abreu MT, Thomas LS, Arnold ET, Lukasek K, Michelsen KS, Arditi M. TLR signaling at the intestinal epithelial interface. J Endotoxin Res. 2003;9:322–330. - PubMed
-
- Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proin-flammatory gene expression in response to bacterial lipopolysac-charide. J Immunol. 2001;167:1609–1616. - PubMed
-
- Akhiani AA, Pappo J, Kabok Z, Schon K, Gao W, Franzen LE, Lycke N. Protection against Helicobacter pylori infection following immunization is IL-12-dependent and mediated by Th1 cells. J Immunol. 2002;169:6977–6984. - PubMed
-
- Akhiani AA, Schon K, Franzen LE, Pappo J, Lycke N. Helicobacter pylori-specific antibodies impair the development of gastritis, facilitate bacterial colonization, and counteract resistance against infection. J Immunol. 2004;172:5024–5033. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical