

Hepatocyte death: a clear and present danger
- PMID: 20664081
- PMCID: PMC2943859
- DOI: 10.1152/physrev.00061.2009
Hepatocyte death: a clear and present danger
Abstract
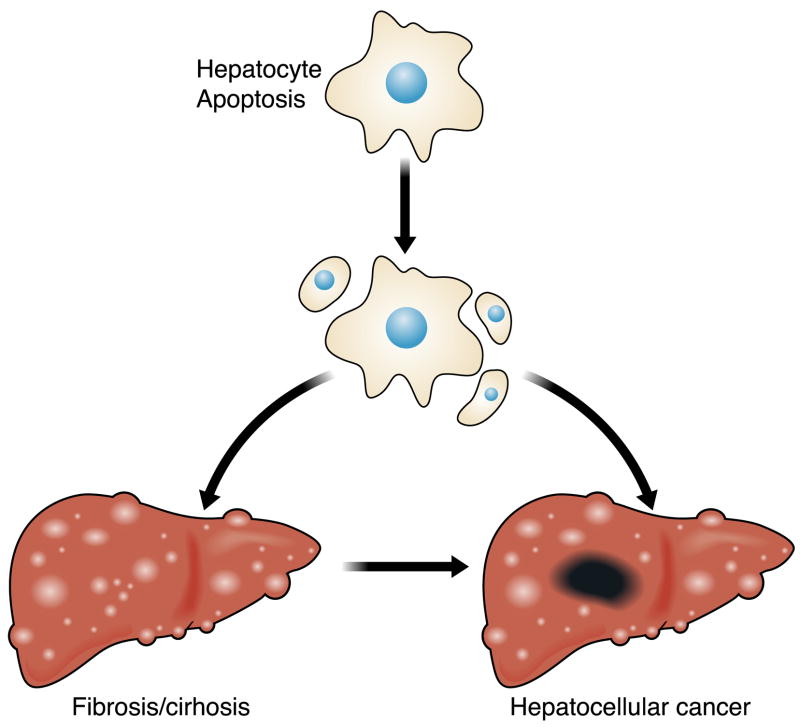
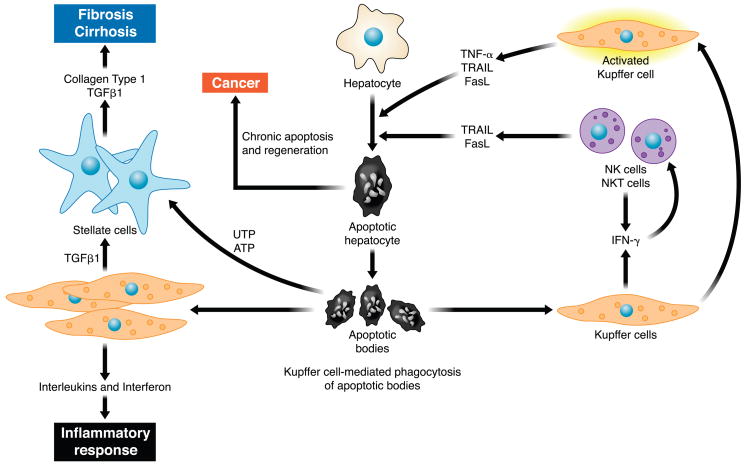
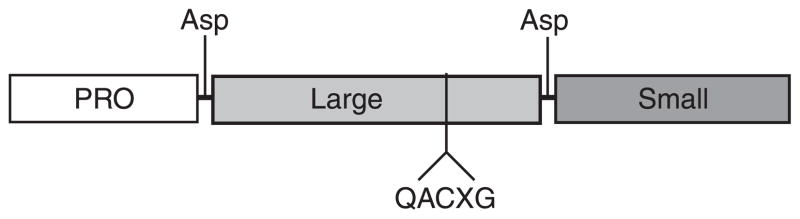
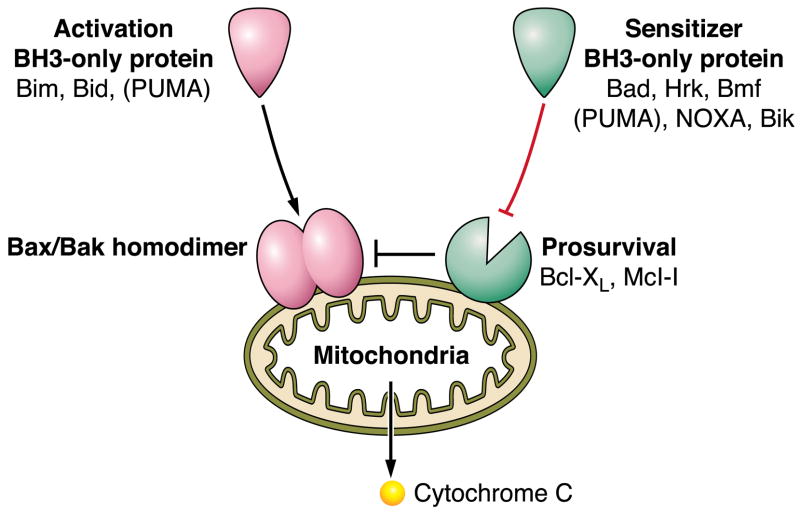
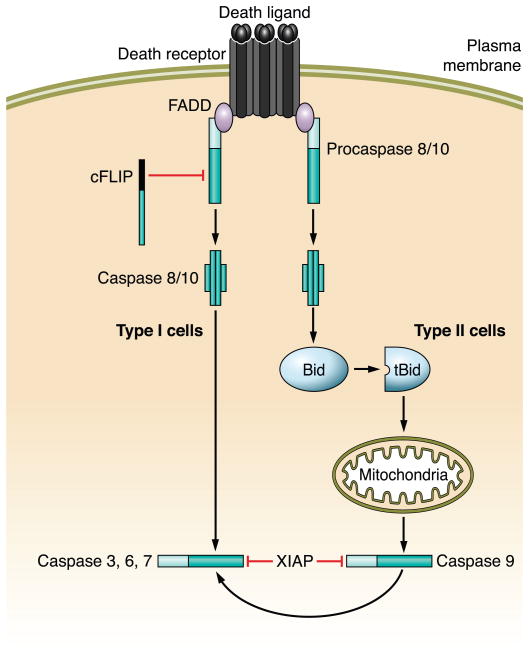
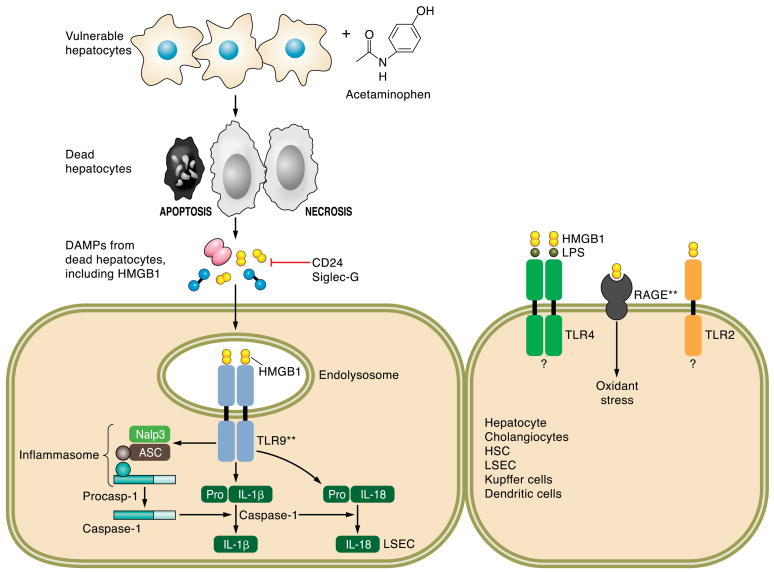
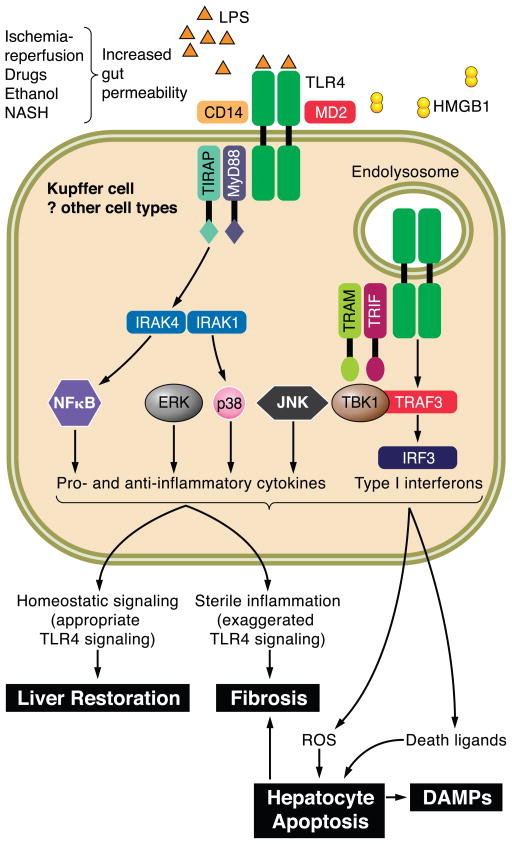
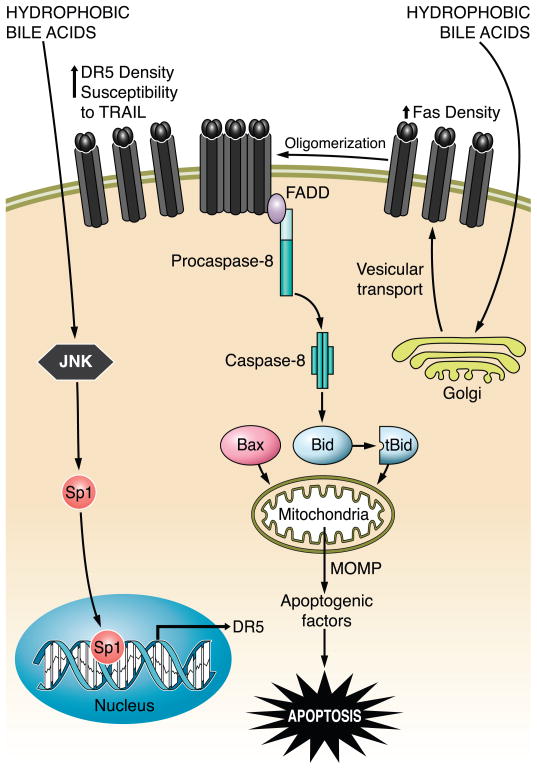
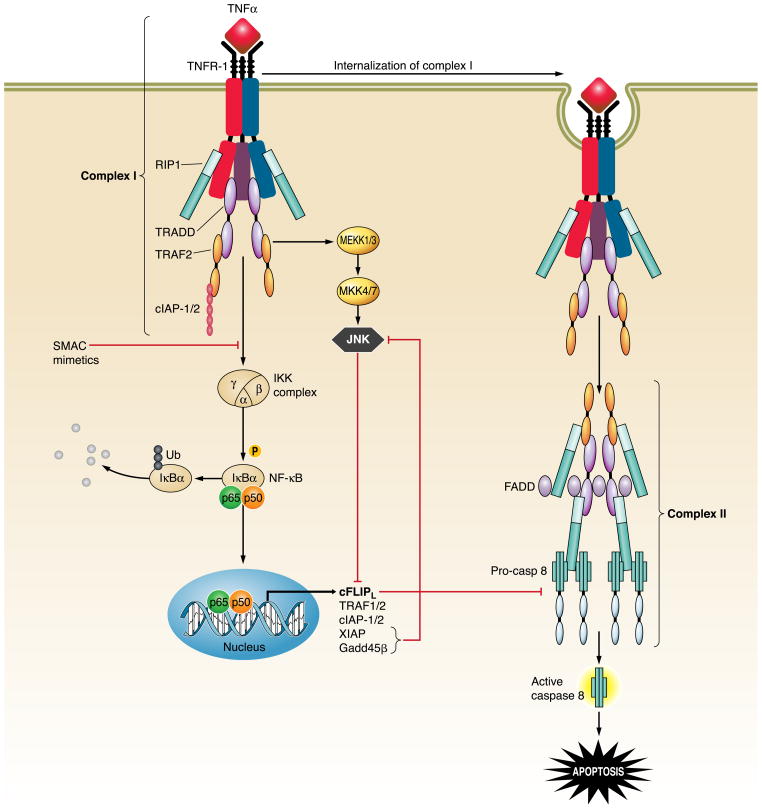
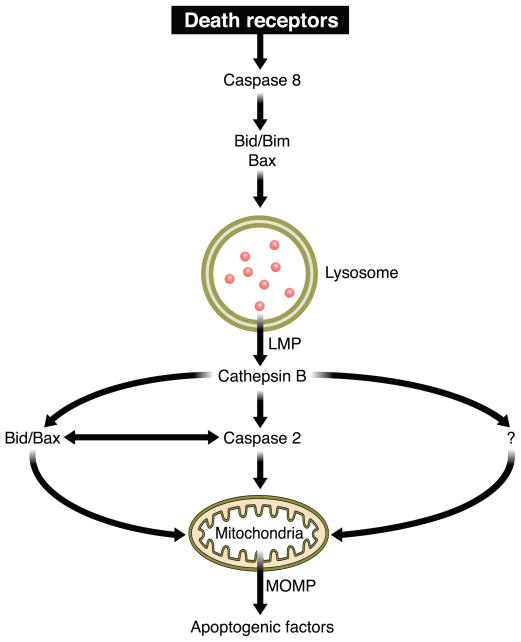
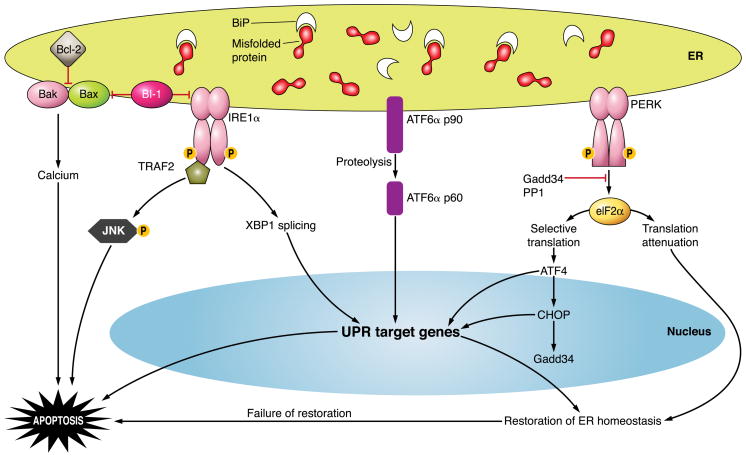
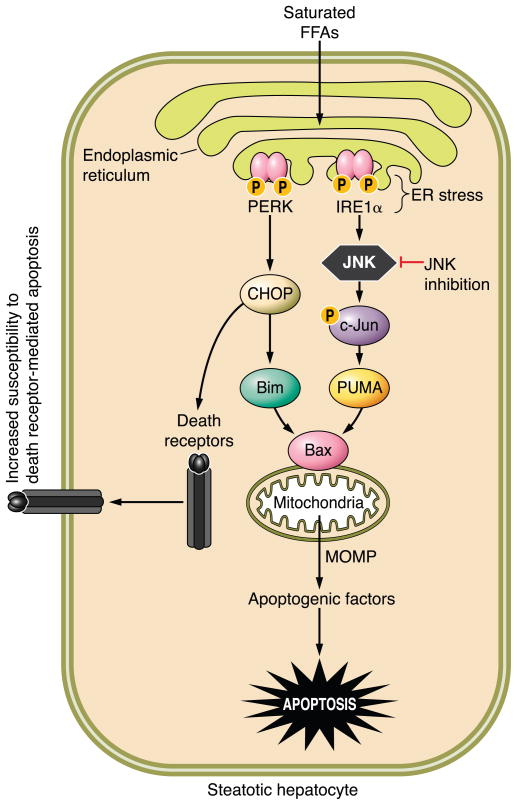
The hepatocyte is especially vulnerable to injury due to its central role in xenobiotic metabolism including drugs and alcohol, participation in lipid and fatty acid metabolism, its unique role in the enterohepatic circulation of bile acids, the widespread prevalence of hepatotropic viruses, and its existence within a milieu of innate immune responding cells. Apoptosis and necrosis are the most widely recognized forms of hepatocyte cell death. The hepatocyte displays many unique features regarding cell death by apoptosis. It is quite susceptible to death receptor-mediated injury, and its death receptor signaling pathways involve the mitochondrial pathway for efficient cell killing. Also, death receptors can trigger lysosomal disruption in hepatocytes which further promote cell and tissue injury. Interestingly, hepatocytes are protected from cell death by only two anti-apoptotic proteins, Bcl-x(L) and Mcl-1, which have nonredundant functions. Endoplasmic reticulum stress or the unfolded protein response contributes to hepatocyte cell death during alterations of lipid and fatty acid metabolism. Finally, the current information implicating RIP kinases in necrosis provides an approach to more fully address this mode of cell death in hepatocyte injury. All of these processes contributing to hepatocyte injury are discussed in the context of potential therapeutic strategies.
Figures
References
-
- Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. - PubMed
-
- Akazawa Y, Mott JL, Bronk SF, Werneburg NW, Kahraman A, Guicciardi ME, Meng XW, Kohno S, Shah VH, Kaufmann SH, McNiven MA, Gores GJ. Death receptor 5 internalization is required for lysosomal permeabilization by TRAIL in malignant liver cell lines. Gastroenterology. 2009;136:2365–2376. - PMC - PubMed
-
- Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n-6/n-3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci. 2004;106:635–643. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous