

Prion protein and Abeta-related synaptic toxicity impairment
- PMID: 20665634
- PMCID: PMC2962809
- DOI: 10.1002/emmm.201000082
Prion protein and Abeta-related synaptic toxicity impairment
Abstract
Alzheimer's disease (AD), the most common neurodegenerative disorder, goes along with extracellular amyloid-beta (Abeta) deposits. The cognitive decline observed during AD progression correlates with damaged spines, dendrites and synapses in hippocampus and cortex. Numerous studies have shown that Abeta oligomers, both synthetic and derived from cultures and AD brains, potently impair synaptic structure and functions. The cellular prion protein (PrP(C)) was proposed to mediate this effect. We report that ablation or overexpression of PrP(C) had no effect on the impairment of hippocampal synaptic plasticity in a transgenic model of AD. These findings challenge the role of PrP(C) as a mediator of Abeta toxicity.
Figures
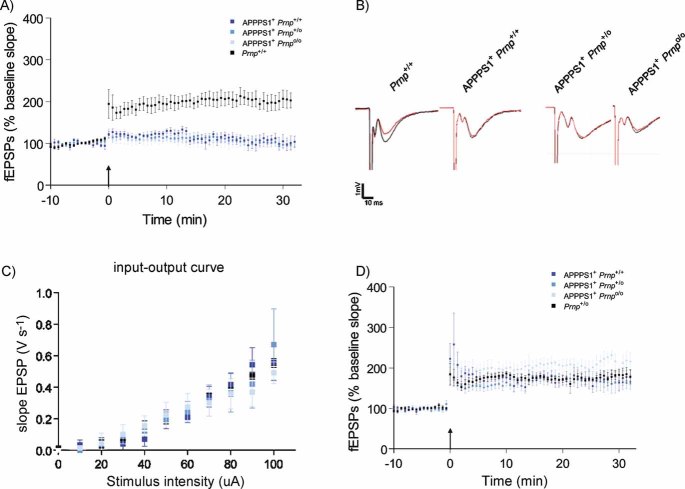
CA1 hippocampal LTP was induced in acute slices from 4-month-old Prnp+/+ mice (black, n = 7), but was abolished in slices from age-matched APPPS1+Prnp+/+ (dark blue, n = 6), APPPS1+Prnp+/o (blue, n = 5) and APPPS1+Prnpo/o mice (light blue n = 5).
fEPSP traces before (red) and after (black) LTP induction. Calibration: 1 mV; 10 ms.
Input–output curves (stimulus intensity vs. fEPSP slope) indicative of normal basal synaptic transmission.
Unaffected LTP in slices derived from 2-month-old APPPS1+Prnp+/+ (n = 5), APPPS1+Prnp+/o (n = 5), APPPS1+Prnpo/o (n = 4) and Prnp+/o mice (n = 5). These results indicate that LTP impairment in APPPS1+ mice was not a developmental defect, and occurred only after 2 months of age independently of Prnp gene dosage.
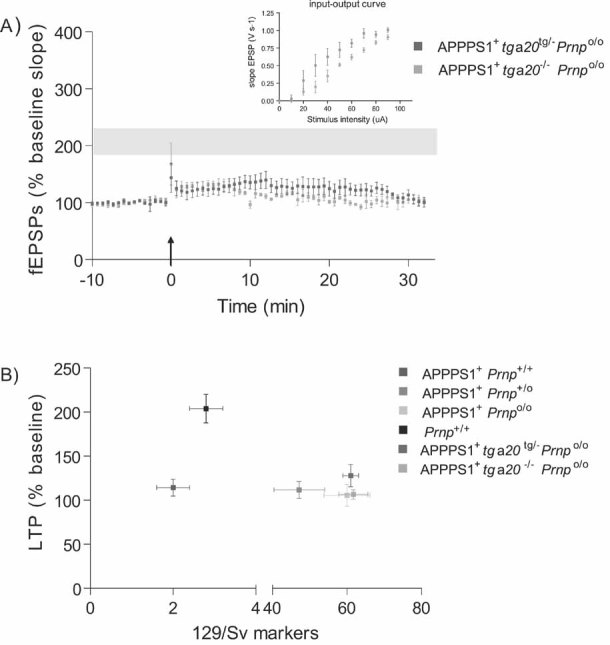
At 4 months of age, LTP was impaired in slices from both APPPS1+tga20tg/−Prnpo/o (n = 4) and APPPS1+tga20−/−Prnpo/o (n = 5) but not in Prnp+/+ slices (n = 7; LTP mean ± SEM from Fig 1A represented as grey ribbon). Basal synaptic transmission was normal as indicated by normal input–output curve (stimulus intensity vs. fEPSP slope).
Average fEPSP slopes (percentage of baseline) at 10–25 min post-LTP plotted against the average number of 129/Sv specific markers for mice depicted in panel A and Fig 1A. In all investigated paradigms, LTP suppression by the APPPS1 transgene was independent of the genetic background.
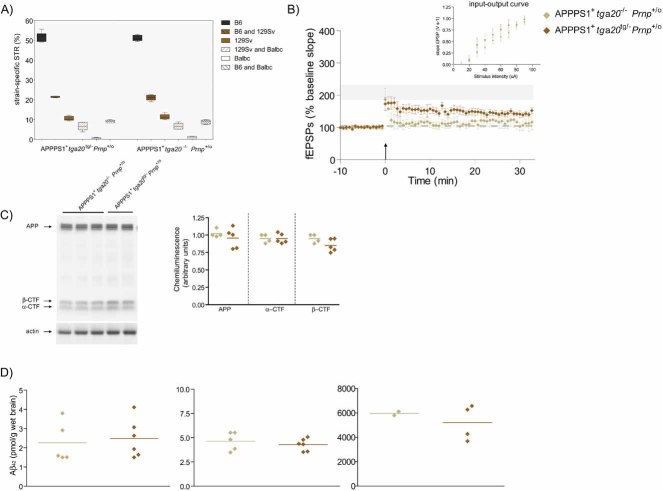
Percentage of strain-specific microsatellites in APPPS1+tga20tg/−Prnp+/o (n = 6) and APPPS1+tga20−/−Prnp+/o (n = 4) mice is displayed by box plot. No significant difference in the genetic background of the two mouse strains was detected (Mann–Whitney U-test, two-tailed, p > 0.05).
At 4 months of age, slices of both APPPS1+tga20tg/−Prnp+/o (n = 6) and APPPS1+tga20−/−Prnp+/o mice (n = 4) displayed reduced LTP when compared to Prnp+/+ mice (n = 7); LTP mean ± SEMfrom Fig 1A represented as grey ribbon. Basal synaptic transmission was normal as indicated by normal input–output curve (stimulus intensity vs. fEPSP slope). All error bars: standard errors of the mean.
APP expression and processing by secretases were similar in 4-month-old APPPS1+ tga20tg/−Prnp+/o and APPPS1+tga20−/−Prnp+/o mice. Left panel: representative SDS–PAGE followed by immunoblotting using an APP C-terminal antibody detecting full-length APP and αβ-CTF; actin was used as loading control. Right panel: quantitation of chemiluminescence for APP, α-CTF and β-CTF.
TRIS-soluble (left panel), detergent-soluble (middle panel) and insoluble (right panel) human Aβ42 levels as assessed by ELISA. Each symbol denotes one individual mouse.
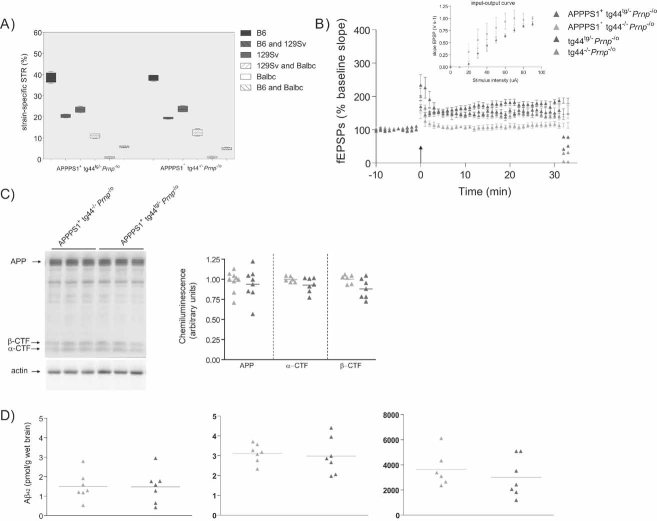
Percentage of strain-specific microsatellites in APPPS1+tg44tg/−Prnp−/o (n = 5) and APPPS1+tg44−/−Prnp−/o (n = 5) mice is displayed by box plot. No significant difference in the genetic background was detected (Mann–Whitney U-test, two-tailed, p > 0.05).
LTP was induced in slices prepared from 4-month-old tg44tg/−Prnp−/o (n = 5) and tg44−/−Prnp−/o (n = 7) mice, but was impaired in slices from APPPS1+tg44−/−Prnp−/o mice (n = 6) and partially rescued in APPPS1+tg44tg/−Prnp−/o (n = 7) mice. Basal synaptic transmission was normal as indicated by normal input–output curve (stimulus intensity vs. fEPSP slope). All mice were compound heterozygotes for the ‘Zurich-I’ (Prnpo) and the ‘Edbg’ (Prnp−) knockout alleles of Prnp.
APP expression and processing by secretases were similar in APPPS1+tg44tg/−Prnp−/o and APPPS1+tg44−/−Prnp−/o mice at 4 months of age. Left panel: representative SDS–PAGE followed by immunoblotting using an APP C-terminal antibody detecting full-length APP and C-terminal fragments (αβ-CTF); actin was used as loading control. Right panel: quantitation of chemiluminescence revealed no difference in APP, α-CTF and β-CTF between the two groups.
TRIS-soluble (left panel), detergent-soluble (middle panel) and insoluble (right panel) human Aβ42 levels as assessed by ELISA. Each symbol denotes one individual mouse.
Comment in
-
Prion protein in Alzheimer's pathogenesis: a hot and controversial issue.EMBO Mol Med. 2010 Aug;2(8):289-90. doi: 10.1002/emmm.201000088. EMBO Mol Med. 2010. PMID: 20698011 Free PMC article.
References
-
- Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13:310–318. - PubMed
-
- Büeler HR, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. - PubMed
-
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
