

Molecular analysis of H7 avian influenza viruses from Australia and New Zealand: genetic diversity and relationships from 1976 to 2007
- PMID: 20668069
- PMCID: PMC2937813
- DOI: 10.1128/JVI.00930-10
Molecular analysis of H7 avian influenza viruses from Australia and New Zealand: genetic diversity and relationships from 1976 to 2007
Abstract
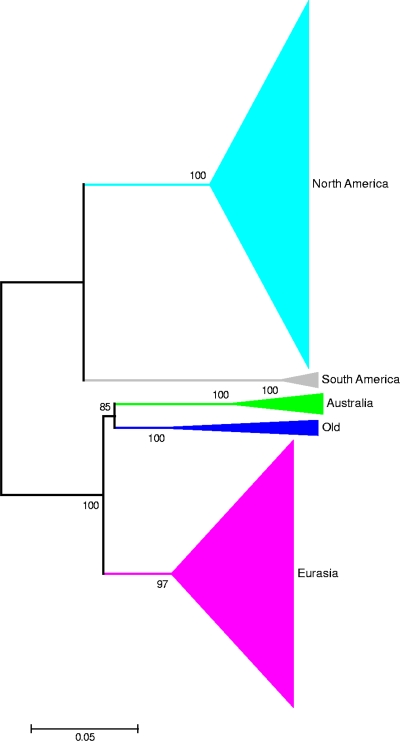
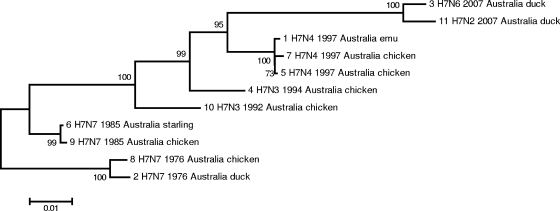
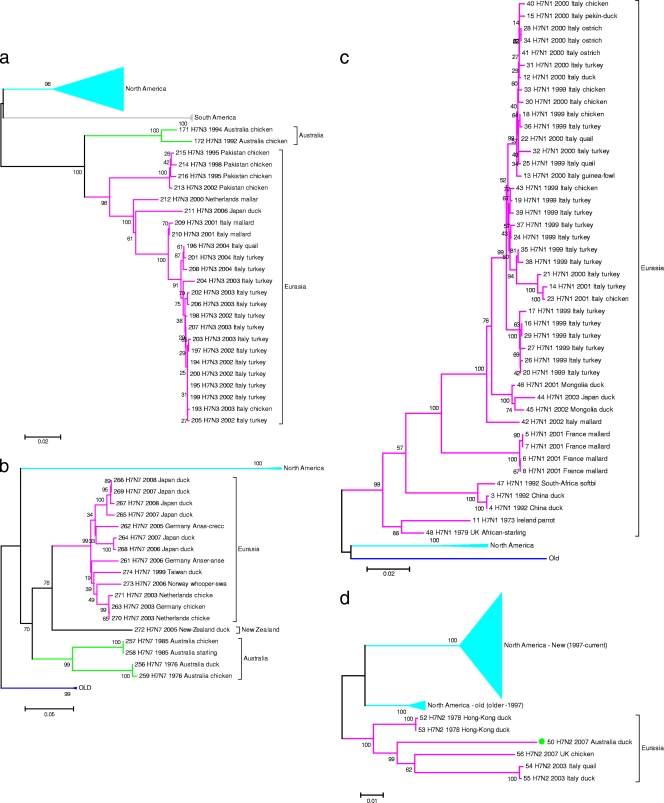
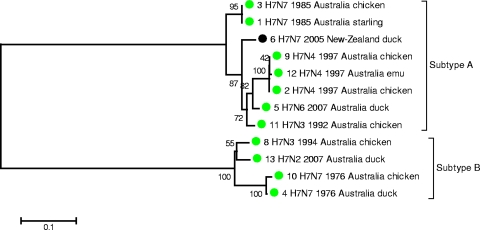
Full-genome sequencing of 11 Australian and 1 New Zealand avian influenza A virus isolate (all subtype H7) has enabled comparison of the sequences of each of the genome segments to those of other subtype H7 avian influenza A viruses. The inference of phylogenetic relationships for each segment has been used to develop a model of the natural history of these viruses in Australia. Phylogenetic analysis of the hemagglutinin segment indicates that the Australian H7 isolates form a monophyletic clade. This pattern is consistent with the long-term, independent evolution that is, in this instance, associated with geographic regions. On the basis of the analysis of the other H7 hemagglutinin sequences, three other geographic regions for which similar monophyletic clades have been observed were confirmed. These regions are Eurasia plus Africa, North America, and South America. Analysis of the neuraminidase sequences from the H7N1, H7N3, and H7N7 genomes revealed the same region-based relationships. This pattern of independent evolution of Australian isolates is supported by the results of analysis of each of the six remaining genomic segments. These results, in conjunction with the occurrence of five different combinations of neuraminidase subtypes (H7N2, H7N3, H7N4, H7N6, H7N7) among the 11 Australian isolates, suggest that the maintenance host(s) is nearly exclusively associated with Australia. The single lineage of Australian H7 hemagglutinin sequences, despite the occurrence of multiple neuraminidase types, suggests the existence of a genetic pool from which a variety of reassortants arise rather than the presence of a small number of stable viral clones. This pattern of evolution is likely to occur in each of the regions mentioned above.
Figures

References
-
- Alexander, D. J. 2003. Should we change the definition of avian influenza for eradication purposes? Avian Dis. 47:976-981. - PubMed
-
- Alexander, D. J., W. H. Allan, D. G. Parsons, and G. Parsons. 1978. Pathogenicity of 4 avian influenza-viruses for fowls, turkeys and ducks. Res. Vet. Sci. 24:242-247. - PubMed
-
- Banks, J., E. C. Speidel, J. W. McCauley, and D. J. Alexander. 2000. Phylogenetic analysis of H7 haemagglutinin subtype influenza A viruses. Arch. Virol. 145:1047-1058. - PubMed
-
- Barr, D. A., A. P. Kelly, R. T. Badman, A. R. Campey, M. D. Orourke, D. C. Grix, and R. L. Reece. 1986. Avian influenza on a multi-age chicken farm. Aust. Vet. J. 63:195-196. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
