

ATP and its N⁶-substituted analogues: parameterization, molecular dynamics simulation and conformational analysis
- PMID: 20668896
- PMCID: PMC3096017
- DOI: 10.1007/s00894-010-0808-3
ATP and its N⁶-substituted analogues: parameterization, molecular dynamics simulation and conformational analysis
Abstract
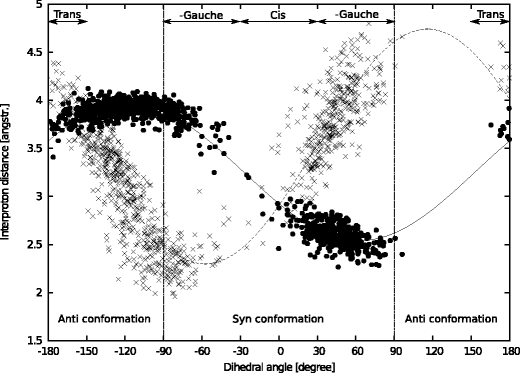
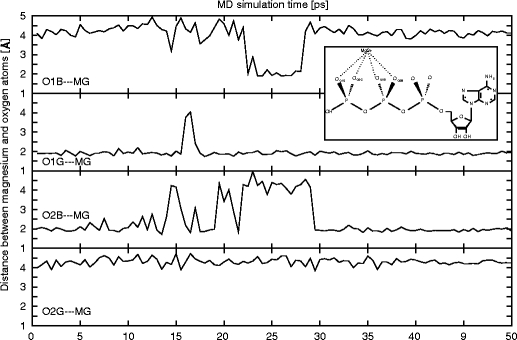
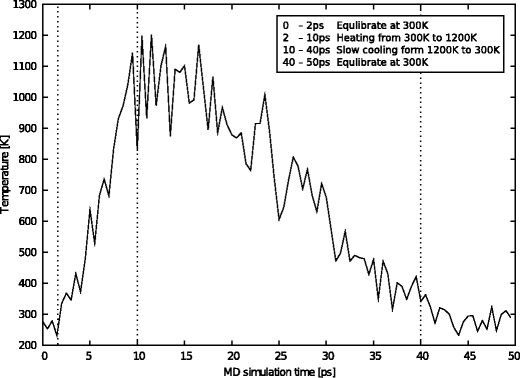
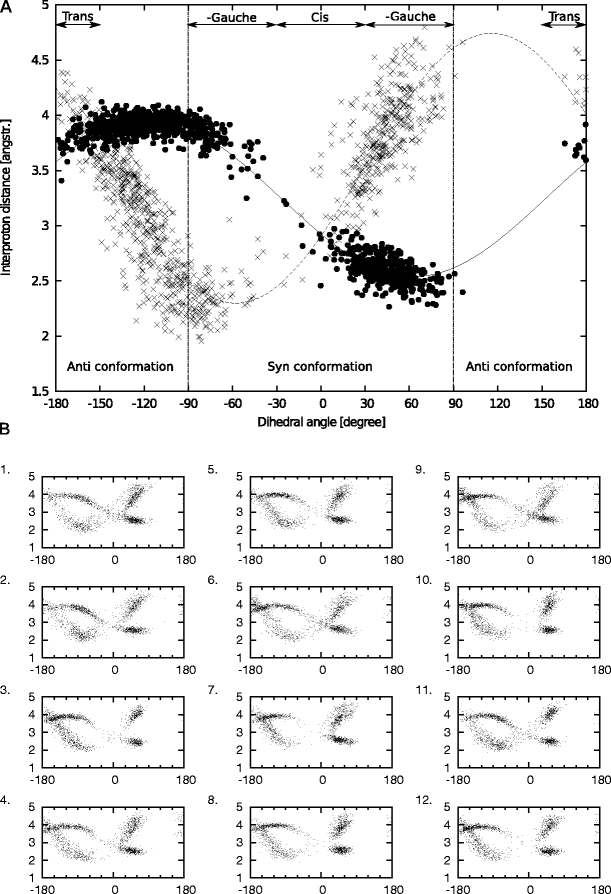
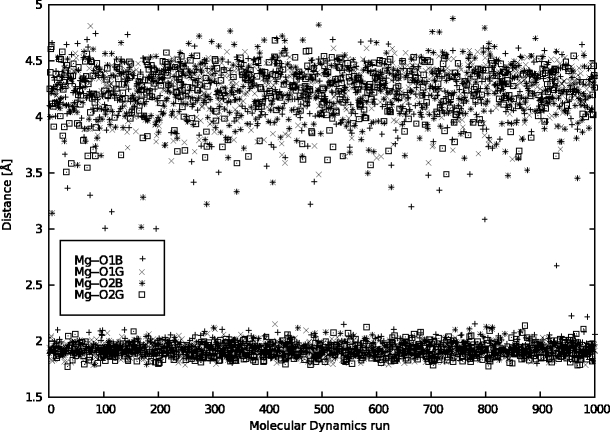
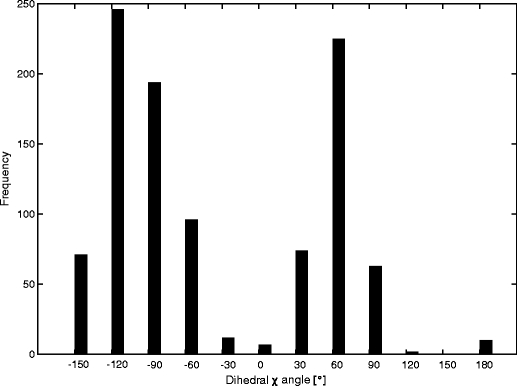
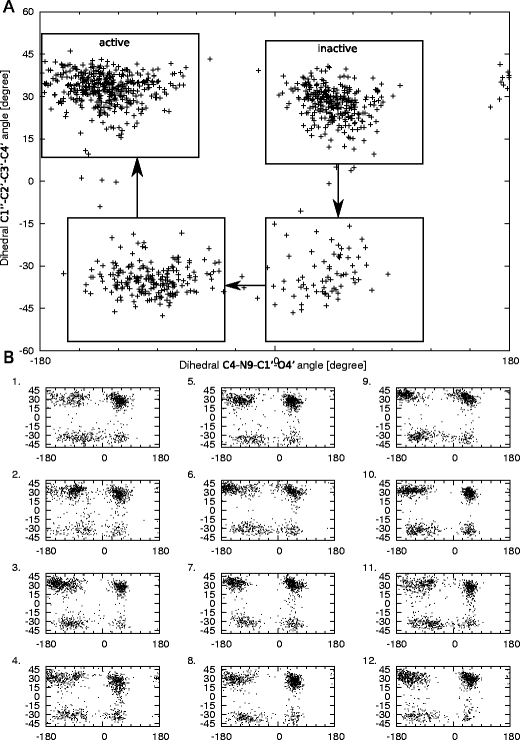
In this work we used a combination of classical molecular dynamics and simulated annealing techniques to shed more light on the conformational flexibility of 12 adenosine triphosphate (ATP) analogues in a water environment. We present simulations in AMBER force field for ATP and 12 published analogues [Shah et al. (1997) Proc Natl Acad Sci USA 94: 3565-3570]. The calculations were carried out using the generalized Born (GB) solvation model in the presence of the cation Mg(2+). The ion was placed at a close distance (2 Å) from the charged oxygen atoms of the beta and gamma phosphate groups of the -3 negatively charged ATP analogue molecules. Analysis of the results revealed the distribution of inter-proton distances H8-H1' and H8-H2' versus the torsion angle ψ (C4-N9-C1'-O4') for all conformations of ATP analogues. There are two gaps in the distribution of torsion angle ψ values: the first is between -30 and 30 degrees and is described by cis-conformation; and the second is between 90 and 175 degrees, which mostly covers a region of anti conformation. Our results compare favorably with results obtained in experimental assays [Jiang and Mao (2002) Polyhedron 21:435-438].
Figures
Similar articles
-
Conformation of ATP and ADP bound to N10-formyltetrahydrofolate synthetase determined by TRNOE NMR spectroscopy.Biochemistry. 1994 Jan 25;33(3):693-8. doi: 10.1021/bi00169a010. Biochemistry. 1994. PMID: 8292596
-
Structural characterization of an N-acetyl-2-aminofluorene (AAF) modified DNA oligomer by NMR, energy minimization, and molecular dynamics.Biochemistry. 1993 Mar 16;32(10):2481-97. doi: 10.1021/bi00061a005. Biochemistry. 1993. PMID: 8448107
-
Conformational dynamics of ATP/Mg:ATP in motor proteins via data mining and molecular simulation.J Chem Phys. 2012 Aug 21;137(7):075101. doi: 10.1063/1.4739308. J Chem Phys. 2012. PMID: 22920142
-
Development of Accurate Force Fields for Mg2+ and Triphosphate Interactions in ATP·Mg2+ and GTP·Mg2+ Complexes.J Chem Theory Comput. 2024 Dec 10;20(23):10553-10563. doi: 10.1021/acs.jctc.4c01142. Epub 2024 Nov 21. J Chem Theory Comput. 2024. PMID: 39571117
-
Structural interpretation of J coupling constants in guanosine and deoxyguanosine: modeling the effects of sugar pucker, backbone conformation, and base pairing.J Phys Chem A. 2009 Jul 23;113(29):8379-86. doi: 10.1021/jp902473v. J Phys Chem A. 2009. PMID: 19569693
References
-
- Burnett G, Kennedy EP. The enzymatic phosphorylation of proteins. J Biol Chem. 1954;211:969–980. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
