

Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags
- PMID: 20673359
- PMCID: PMC2921404
- DOI: 10.1186/1471-2180-10-206
Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags
Abstract
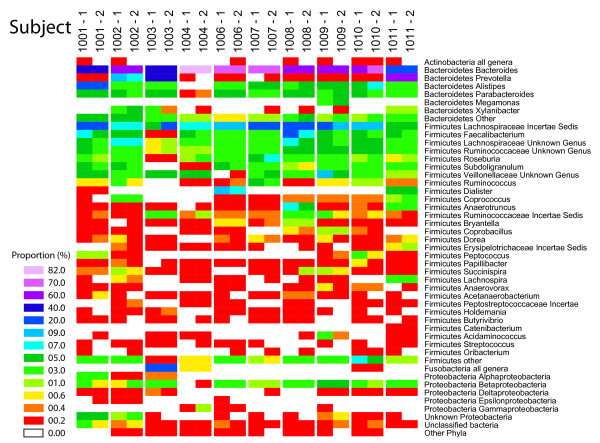
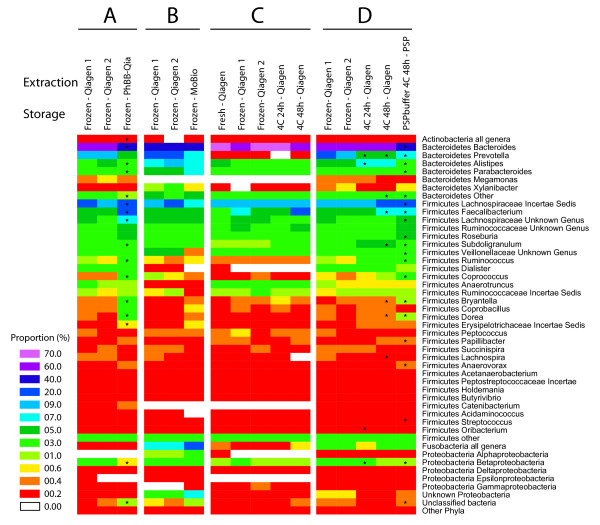
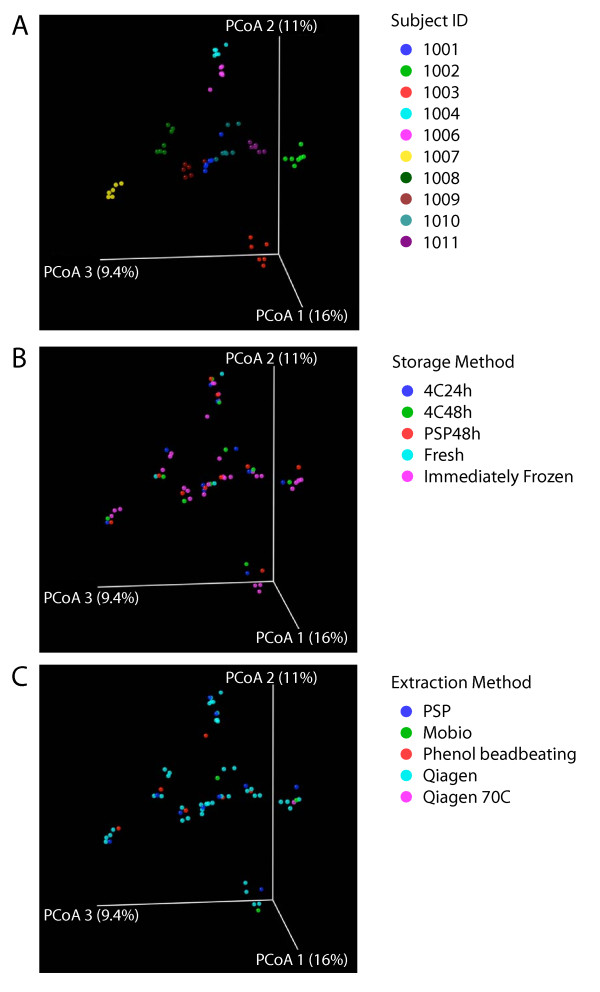
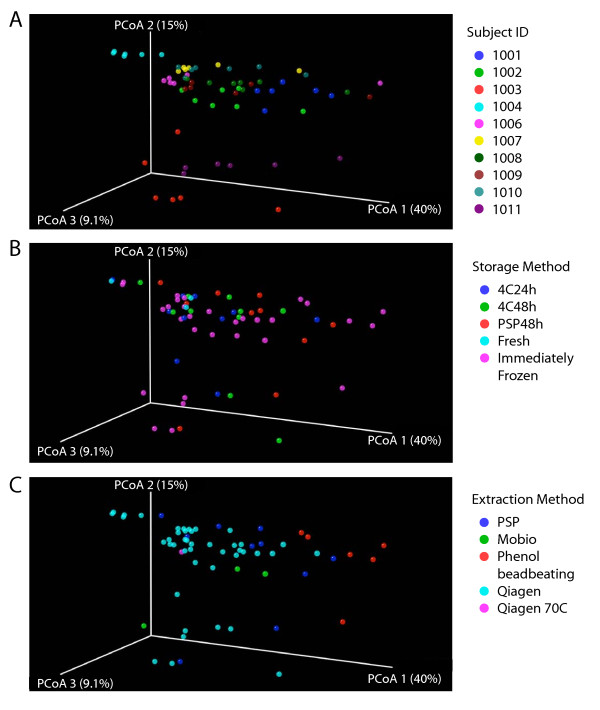
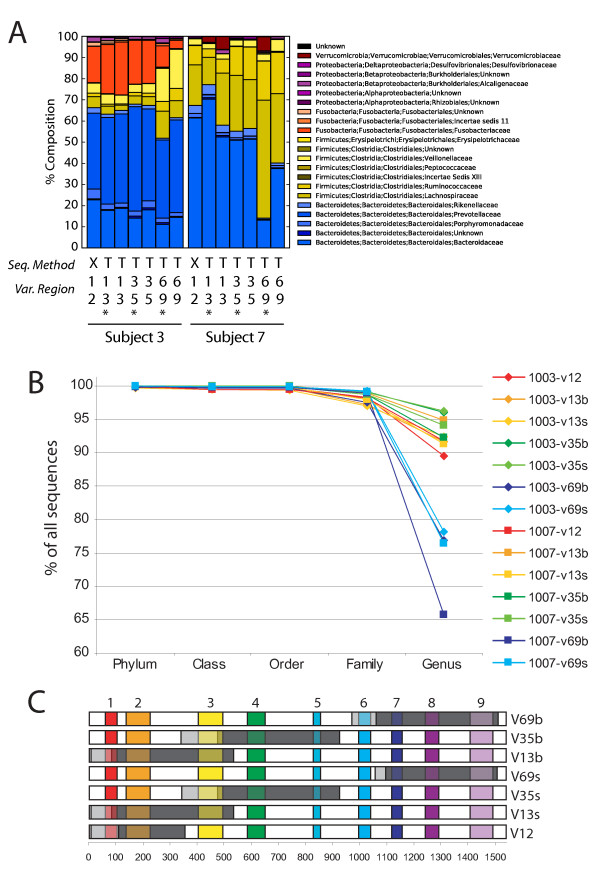
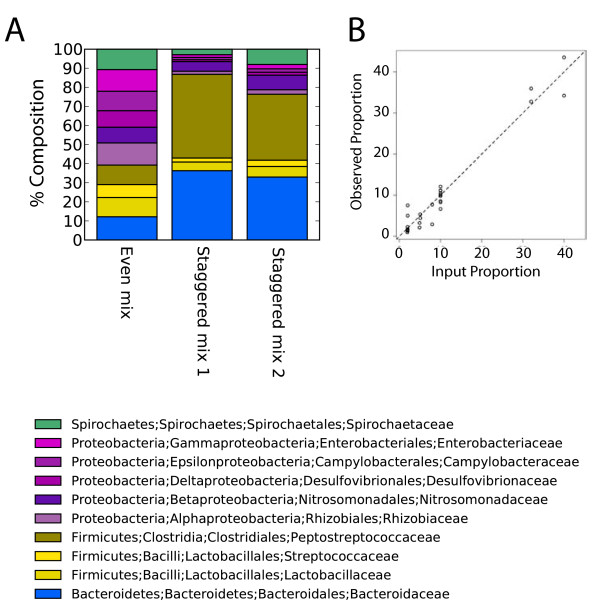
Intense interest centers on the role of the human gut microbiome in health and disease, but optimal methods for analysis are still under development. Here we present a study of methods for surveying bacterial communities in human feces using 454/Roche pyrosequencing of 16S rRNA gene tags. We analyzed fecal samples from 10 individuals and compared methods for storage, DNA purification and sequence acquisition. To assess reproducibility, we compared samples one cm apart on a single stool specimen for each individual. To analyze storage methods, we compared 1) immediate freezing at -80 degrees C, 2) storage on ice for 24 or 3) 48 hours. For DNA purification methods, we tested three commercial kits and bead beating in hot phenol. Variations due to the different methodologies were compared to variation among individuals using two approaches--one based on presence-absence information for bacterial taxa (unweighted UniFrac) and the other taking into account their relative abundance (weighted UniFrac). In the unweighted analysis relatively little variation was associated with the different analytical procedures, and variation between individuals predominated. In the weighted analysis considerable variation was associated with the purification methods. Particularly notable was improved recovery of Firmicutes sequences using the hot phenol method. We also carried out surveys of the effects of different 454 sequencing methods (FLX versus Titanium) and amplification of different 16S rRNA variable gene segments. Based on our findings we present recommendations for protocols to collect, process and sequence bacterial 16S rDNA from fecal samples--some major points are 1) if feasible, bead-beating in hot phenol or use of the PSP kit improves recovery; 2) storage methods can be adjusted based on experimental convenience; 3) unweighted (presence-absence) comparisons are less affected by lysis method.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
