

Characterization of two Runx1-dependent nociceptor differentiation programs necessary for inflammatory versus neuropathic pain
- PMID: 20673362
- PMCID: PMC2919460
- DOI: 10.1186/1744-8069-6-45
Characterization of two Runx1-dependent nociceptor differentiation programs necessary for inflammatory versus neuropathic pain
Abstract
Background: The cellular and molecular programs that control specific types of pain are poorly understood. We reported previously that the runt domain transcription factor Runx1 is initially expressed in most nociceptors and controls sensory neuron phenotypes necessary for inflammatory and neuropathic pain.
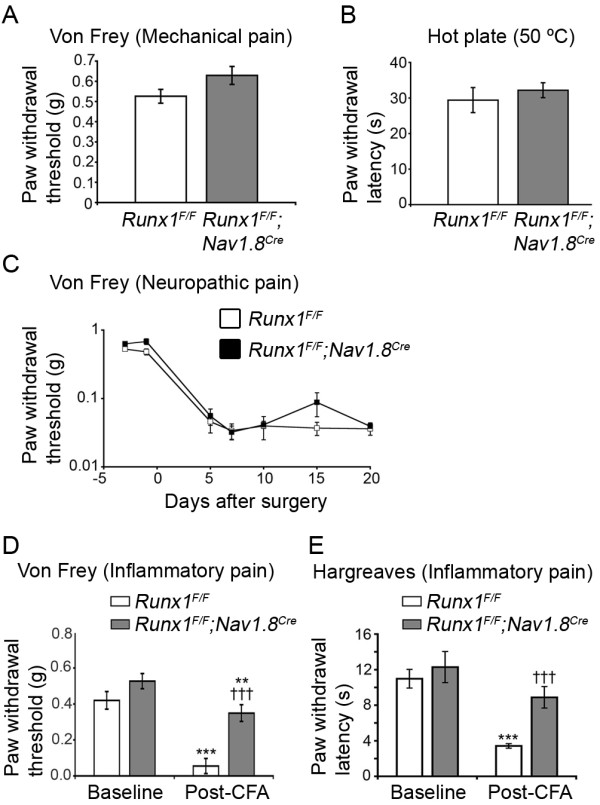
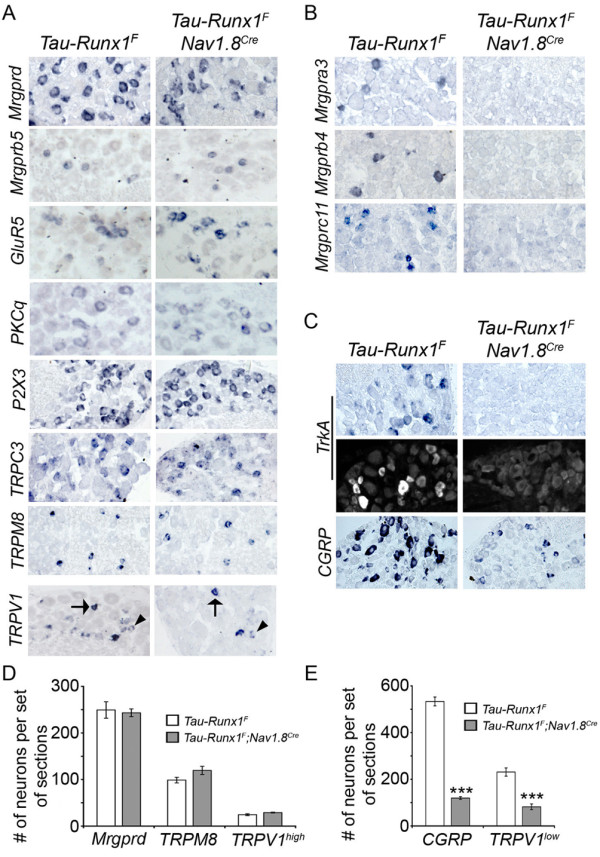
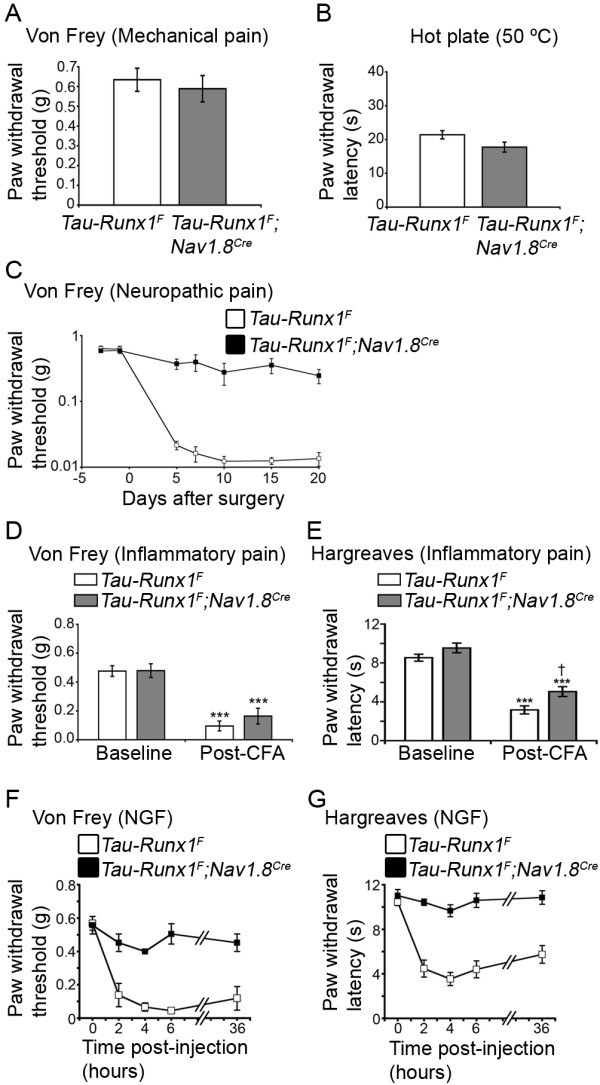
Results: Here we show that expression of Runx1-dependent ion channels and receptors is distributed into two nociceptor populations that are distinguished by persistent or transient Runx1 expression. Conditional mutation of Runx1 at perinatal stages leads to preferential impairment of Runx1-persistent nociceptors and a selective defect in inflammatory pain. Conversely, constitutive Runx1 expression in Runx1-transient nociceptors leads to an impairment of Runx1-transient nociceptors and a selective deficit in neuropathic pain. Notably, the subdivision of Runx1-persistent and Runx1-transient nociceptors does not follow the classical nociceptor subdivision into IB4+ nonpeptidergic and IB4- peptidergic populations.
Conclusion: Altogether, we have uncovered two distinct Runx1-dependent nociceptor differentiation programs that are permissive for inflammatory versus neuropathic pain. These studies lend support to a transcription factor-based distinction of neuronal classes necessary for inflammatory versus neuropathic pain.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
