

Mutations in IMPG2, encoding interphotoreceptor matrix proteoglycan 2, cause autosomal-recessive retinitis pigmentosa
- PMID: 20673862
- PMCID: PMC2917719
- DOI: 10.1016/j.ajhg.2010.07.004
Mutations in IMPG2, encoding interphotoreceptor matrix proteoglycan 2, cause autosomal-recessive retinitis pigmentosa
Abstract
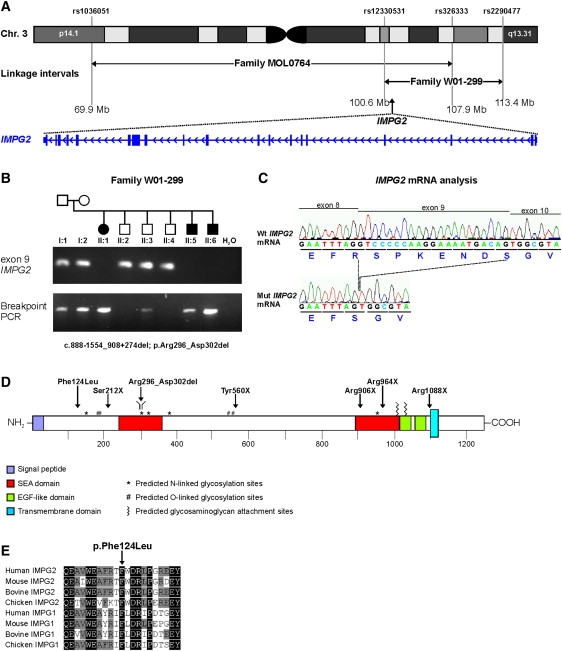
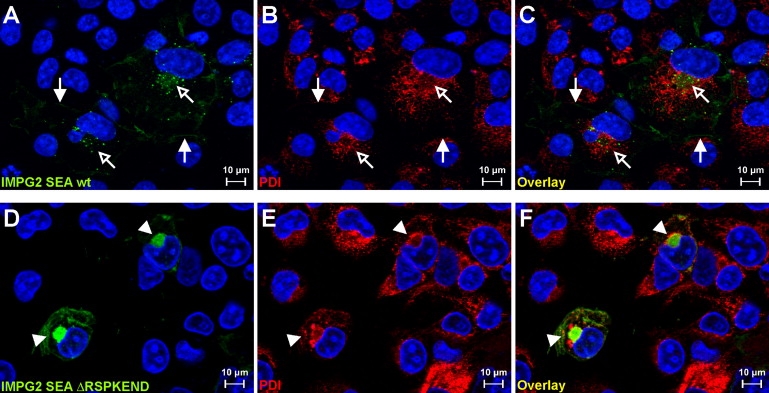
Retinitis pigmentosa (RP) is a heterogeneous group of inherited retinal diseases caused by progressive degeneration of the photoreceptor cells. Using autozygosity mapping, we identified two families, each with three affected siblings sharing large overlapping homozygous regions that harbored the IMPG2 gene on chromosome 3. Sequence analysis of IMPG2 in the two index cases revealed homozygous mutations cosegregating with the disease in the respective families: three affected siblings of Iraqi Jewish ancestry displayed a nonsense mutation, and a Dutch family displayed a 1.8 kb genomic deletion that removes exon 9 and results in the absence of seven amino acids in a conserved SEA domain of the IMPG2 protein. Transient transfection of COS-1 cells showed that a construct expressing the wild-type SEA domain is properly targeted to the plasma membrane, whereas the mutant lacking the seven amino acids appears to be retained in the endoplasmic reticulum. Mutation analysis in ten additional index cases that were of Dutch, Israeli, Italian, and Pakistani origin and had homozygous regions encompassing IMPG2 revealed five additional mutations; four nonsense mutations and one missense mutation affecting a highly conserved phenylalanine residue. Most patients with IMPG2 mutations showed an early-onset form of RP with progressive visual-field loss and deterioration of visual acuity. The patient with the missense mutation, however, was diagnosed with maculopathy. The IMPG2 gene encodes the interphotoreceptor matrix proteoglycan IMPG2, which is a constituent of the interphotoreceptor matrix. Our data therefore show that mutations in a structural component of the interphotoreceptor matrix can cause arRP.
Figures


Similar articles
-
IMPG2-associated retinitis pigmentosa displays relatively early macular involvement.Invest Ophthalmol Vis Sci. 2014 May 29;55(6):3939-53. doi: 10.1167/iovs.14-14129. Invest Ophthalmol Vis Sci. 2014. PMID: 24876279
-
A mutation in ZNF513, a putative regulator of photoreceptor development, causes autosomal-recessive retinitis pigmentosa.Am J Hum Genet. 2010 Sep 10;87(3):400-9. doi: 10.1016/j.ajhg.2010.08.003. Am J Hum Genet. 2010. PMID: 20797688 Free PMC article.
-
Missense mutations at homologous positions in the fourth and fifth laminin A G-like domains of eyes shut homolog cause autosomal recessive retinitis pigmentosa.Mol Vis. 2010 Dec 15;16:2753-9. Mol Vis. 2010. PMID: 21179430 Free PMC article.
-
Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa.Am J Hum Genet. 2010 Sep 10;87(3):382-91. doi: 10.1016/j.ajhg.2010.07.022. Epub 2010 Aug 12. Am J Hum Genet. 2010. PMID: 20705279 Free PMC article.
-
A novel homozygous R764H mutation in crumbs homolog 1 causes autosomal recessive retinitis pigmentosa.Mol Vis. 2013 Apr 5;19:829-34. Print 2013. Mol Vis. 2013. PMID: 23592920 Free PMC article.
Cited by
-
Retinal characteristics of the congenital disorder of glycosylation PMM2-CDG.J Inherit Metab Dis. 2013 Nov;36(6):1039-47. doi: 10.1007/s10545-013-9594-2. Epub 2013 Feb 22. J Inherit Metab Dis. 2013. PMID: 23430200
-
Clinical and genetic investigations in Chinese families with retinitis pigmentosa.Exp Biol Med (Maywood). 2022 Jun;247(12):1030-1038. doi: 10.1177/15353702221085711. Epub 2022 Apr 11. Exp Biol Med (Maywood). 2022. PMID: 35410501 Free PMC article.
-
Proteoglycan IMPG2 Shapes the Interphotoreceptor Matrix and Modulates Vision.J Neurosci. 2020 May 13;40(20):4059-4072. doi: 10.1523/JNEUROSCI.2994-19.2020. Epub 2020 Apr 7. J Neurosci. 2020. PMID: 32265257 Free PMC article.
-
The molecular basis of retinal dystrophies in pakistan.Genes (Basel). 2014 Mar 11;5(1):176-95. doi: 10.3390/genes5010176. Genes (Basel). 2014. PMID: 24705292 Free PMC article.
-
A novel DFNB31 mutation associated with Usher type 2 syndrome showing variable degrees of auditory loss in a consanguineous Portuguese family.Mol Vis. 2011;17:1598-606. Epub 2011 Jun 15. Mol Vis. 2011. PMID: 21738389 Free PMC article.
References
-
- Bunker C.H., Berson E.L., Bromley W.C., Hayes R.P., Roderick T.H. Prevalence of retinitis pigmentosa in Maine. Am. J. Ophthalmol. 1984;97:357–365. - PubMed
-
- Rosenberg T. Epidemiology of hereditary ocular disorders. Dev. Ophthalmol. 2003;37:16–33. - PubMed
-
- Berson E.L. Retinitis pigmentosa. The Friedenwald Lecture. Invest. Ophthalmol. Vis. Sci. 1993;34:1659–1676. - PubMed
-
- Hartong D.T., Berson E.L., Dryja T.P. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
