

Editorial
doi: 10.3324/haematol.2010.025619.
Inherited bone marrow failure syndromes
- PMID: 20675743
- PMCID: PMC2913069
- DOI: 10.3324/haematol.2010.025619
Item in Clipboard
Editorial
Inherited bone marrow failure syndromes
Haematologica.
2010 Aug.
No abstract available
Figures
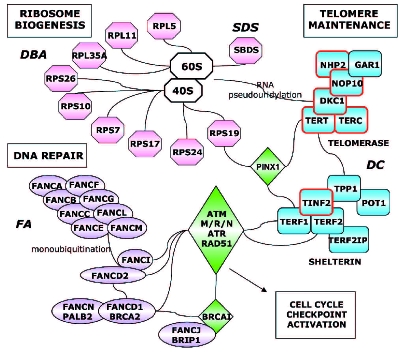
Interconnected pathways that cause bone marrow failure. Genes known to be mutated in three different pathways that lead to bone marrow failure are highlighted. Genes mutated in the telomere maintenance pathway are circled in red. Interactions are as defined in Entrez Gene at http://www.ncbi.nlm.nih.gov/ . Gene names for those mutated as well as for several key interconnecting proteins are as follows: RP ribosomal protein; FANC, Fanconi anemia complementation group; DKC1, dyskeratosis congenita 1, dyskerin; NOP10, nucleolar protein 10 homolog; NHP2, non-histone ribonucleoprotein 2 homolog; GAR1, glycine and arginine rich ribonucleoprotein 1 homolog; TERF1, telomeric repeat binding factor 1; TERF2, telomeric repeat binding factor 2; TINF2, TERF1-interacting nuclear factor 2; TERF2IP, TERF2 interacting protein (RAP1); POT1 protection of telomeres 1 homolog; TPP1, TIN2 interacting protein 1 (= ACD, adrenocortical dysplasia homolog); PINX1, PIN2 (=TERF1) interacting protein; ATM, ataxia telangiectasia mutated; M/R/N = MRE11/RAD50/NBS1, meiotic recombination 11 homolog A/radiation resistance 50 homolog/Nijmegen breakage syndrome 1; ATR, ataxia telangiectasia and Rad3 related (Seckel syndrome); BRCA1, breast cancer 1. BRIP1, BRCA1 interacting protein C-terminal helicase 1; PALB2, partner and localizer of BRCA2 (modified from reference 20).
Comment on
-
Mutations in the ribosomal protein genes in Japanese patients with Diamond-Blackfan anemia.Haematologica. 2010 Aug;95(8):1293-9. doi: 10.3324/haematol.2009.020826. Epub 2010 Apr 7. Haematologica. 2010. PMID: 20378560 Free PMC article.
-
Frequency and natural history of inherited bone marrow failure syndromes: the Israeli Inherited Bone Marrow Failure Registry.Haematologica. 2010 Aug;95(8):1300-7. doi: 10.3324/haematol.2009.018119. Epub 2010 Apr 30. Haematologica. 2010. PMID: 20435624 Free PMC article.
References
-
- Auerbach AD, Buchwald M, Joenje H. In: The Metabolic and Molecular Basis Of Inherited Disease. Scriver CR, et al., editors. McGraw-Hill; New York: 2001. pp. 753–68.
-
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De DieSmulders C, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297(5581):606–9. - PubMed
-
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8(10):735–48. - PubMed
-
- Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nature Genet. 1998;19(1):32–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
