

A comparison of batch effect removal methods for enhancement of prediction performance using MAQC-II microarray gene expression data
- PMID: 20676067
- PMCID: PMC2920074
- DOI: 10.1038/tpj.2010.57
A comparison of batch effect removal methods for enhancement of prediction performance using MAQC-II microarray gene expression data
Abstract
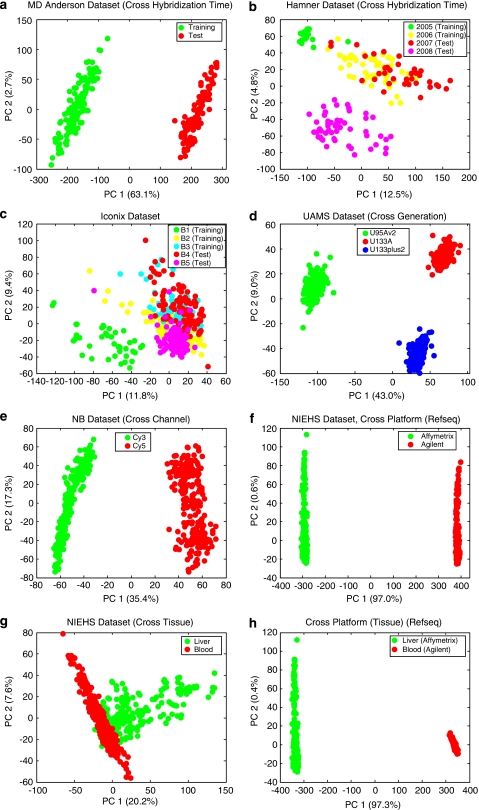
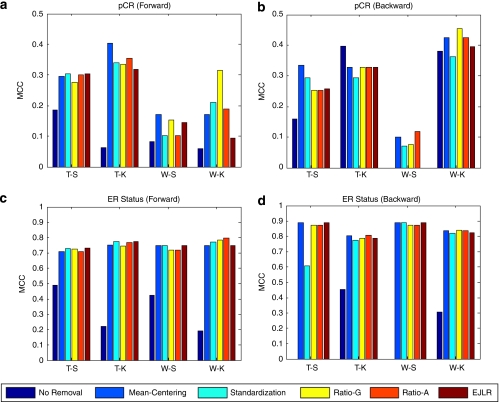
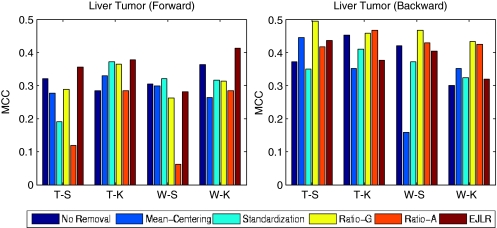
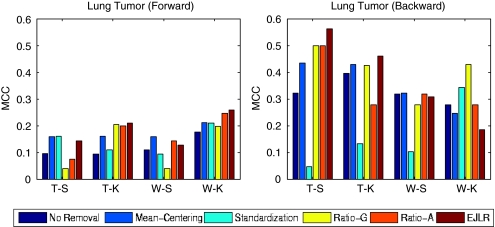
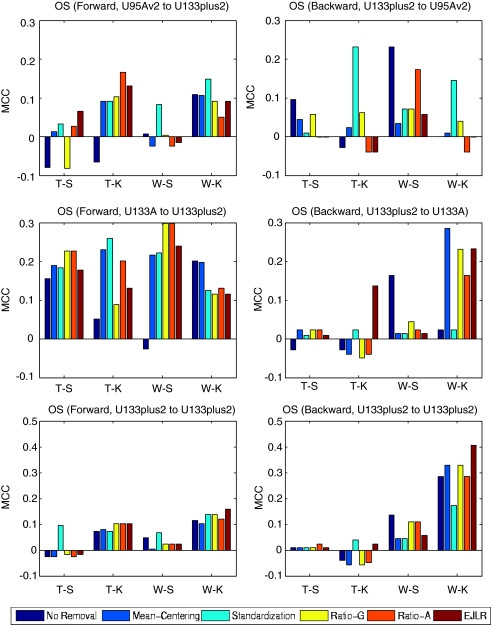
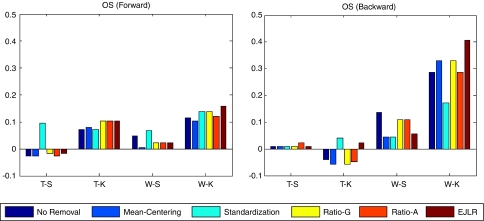
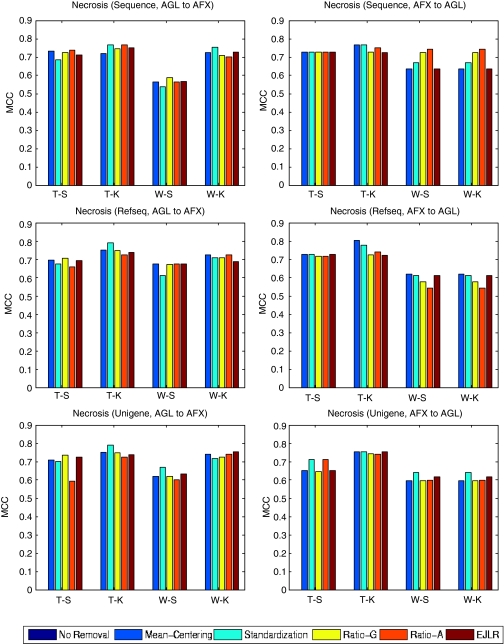
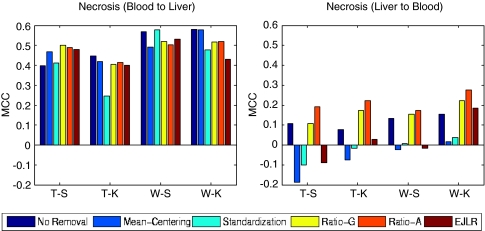
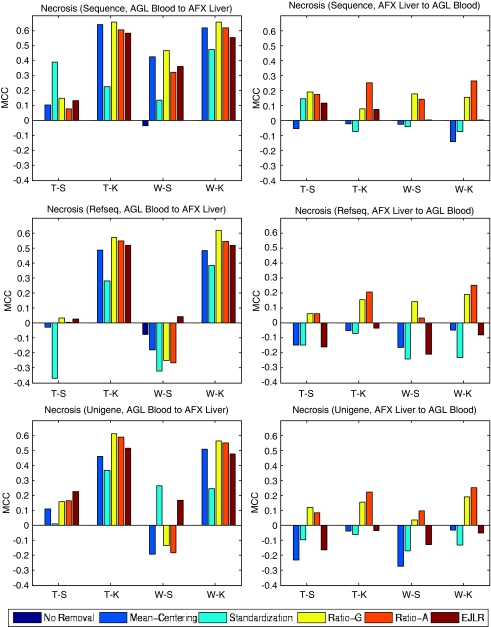
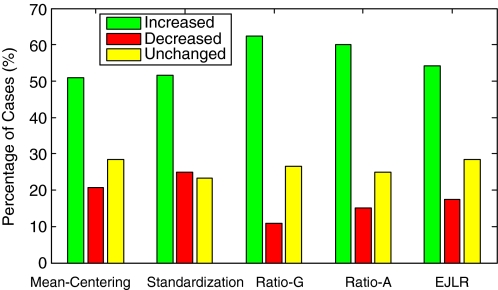
Batch effects are the systematic non-biological differences between batches (groups) of samples in microarray experiments due to various causes such as differences in sample preparation and hybridization protocols. Previous work focused mainly on the development of methods for effective batch effects removal. However, their impact on cross-batch prediction performance, which is one of the most important goals in microarray-based applications, has not been addressed. This paper uses a broad selection of data sets from the Microarray Quality Control Phase II (MAQC-II) effort, generated on three microarray platforms with different causes of batch effects to assess the efficacy of their removal. Two data sets from cross-tissue and cross-platform experiments are also included. Of the 120 cases studied using Support vector machines (SVM) and K nearest neighbors (KNN) as classifiers and Matthews correlation coefficient (MCC) as performance metric, we find that Ratio-G, Ratio-A, EJLR, mean-centering and standardization methods perform better or equivalent to no batch effect removal in 89, 85, 83, 79 and 75% of the cases, respectively, suggesting that the application of these methods is generally advisable and ratio-based methods are preferred.
Figures
References
-
- Affymetrix Microarray Suite User GuideVersion 5. Affymetrix2001
-
- Irizarry RA, Hobbs B, Collin F, Beazer-barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. - PubMed
-
- Li C, Wing H.DNA-Chip Analyzer (dChip). The analysis of gene expression data: methods and softwareG Parmigiani, ES Garrett, R Irizarry and SL Zeger (eds).Springer, New York; 2003120–141.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
