

p66(Shc) restrains Ras hyperactivation and suppresses metastatic behavior
- PMID: 20676142
- PMCID: PMC3045677
- DOI: 10.1038/onc.2010.326
p66(Shc) restrains Ras hyperactivation and suppresses metastatic behavior
Abstract
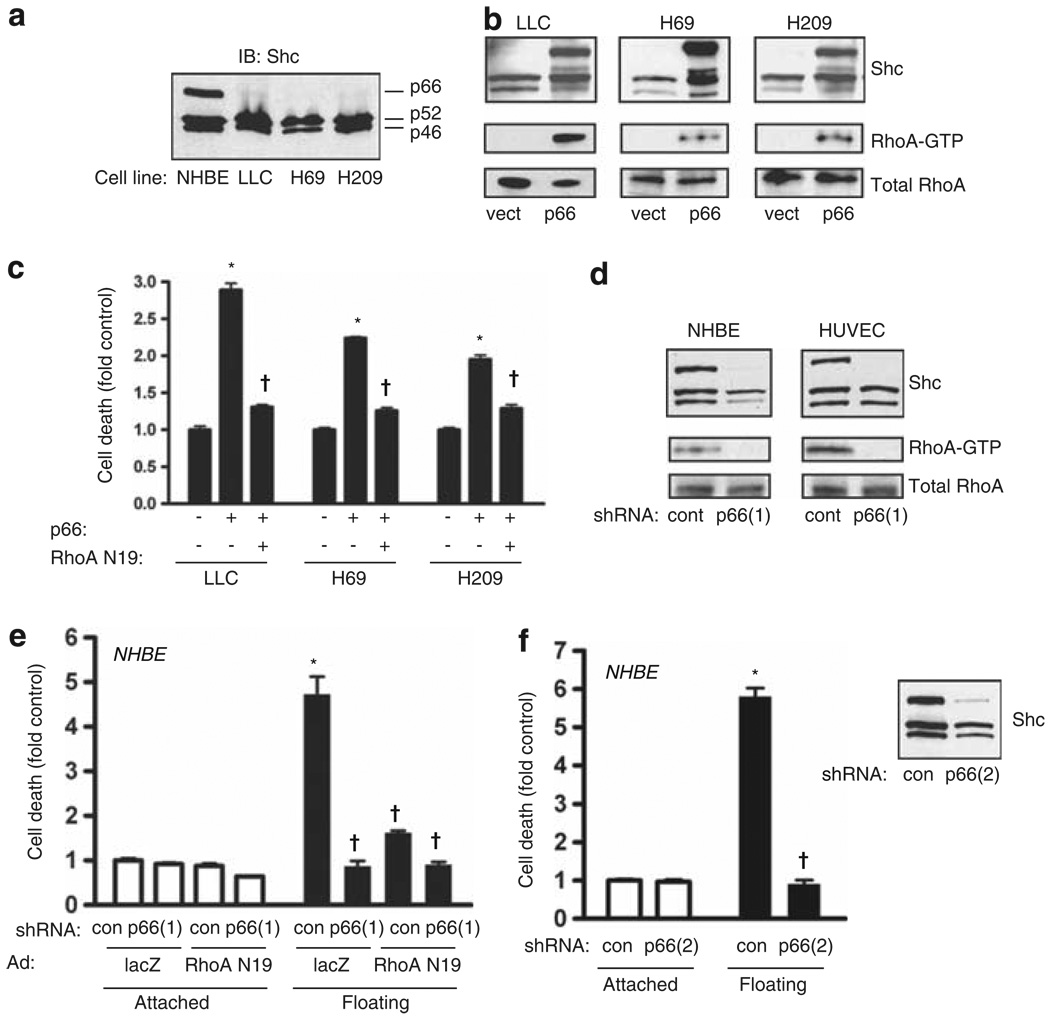
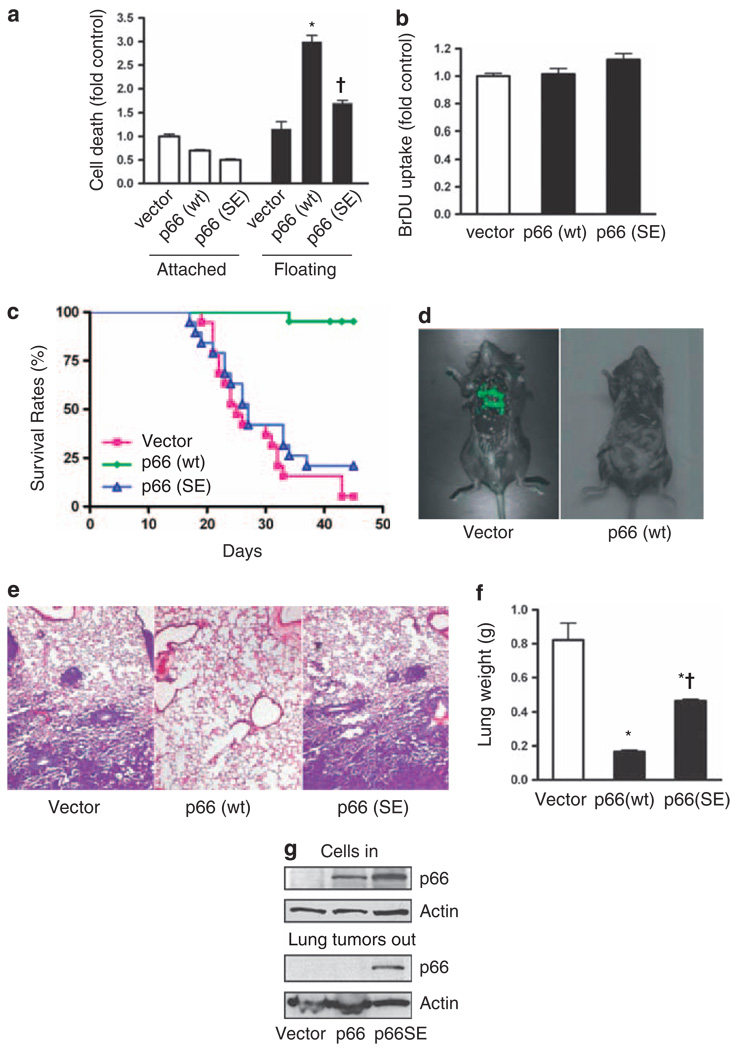
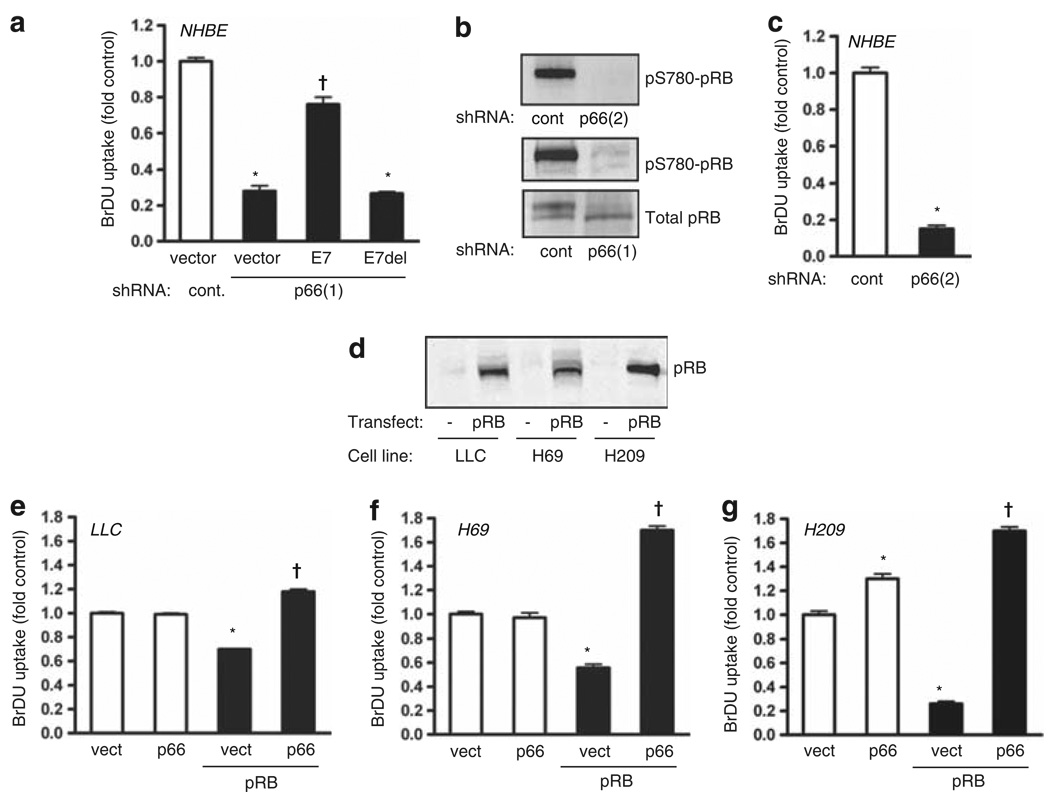
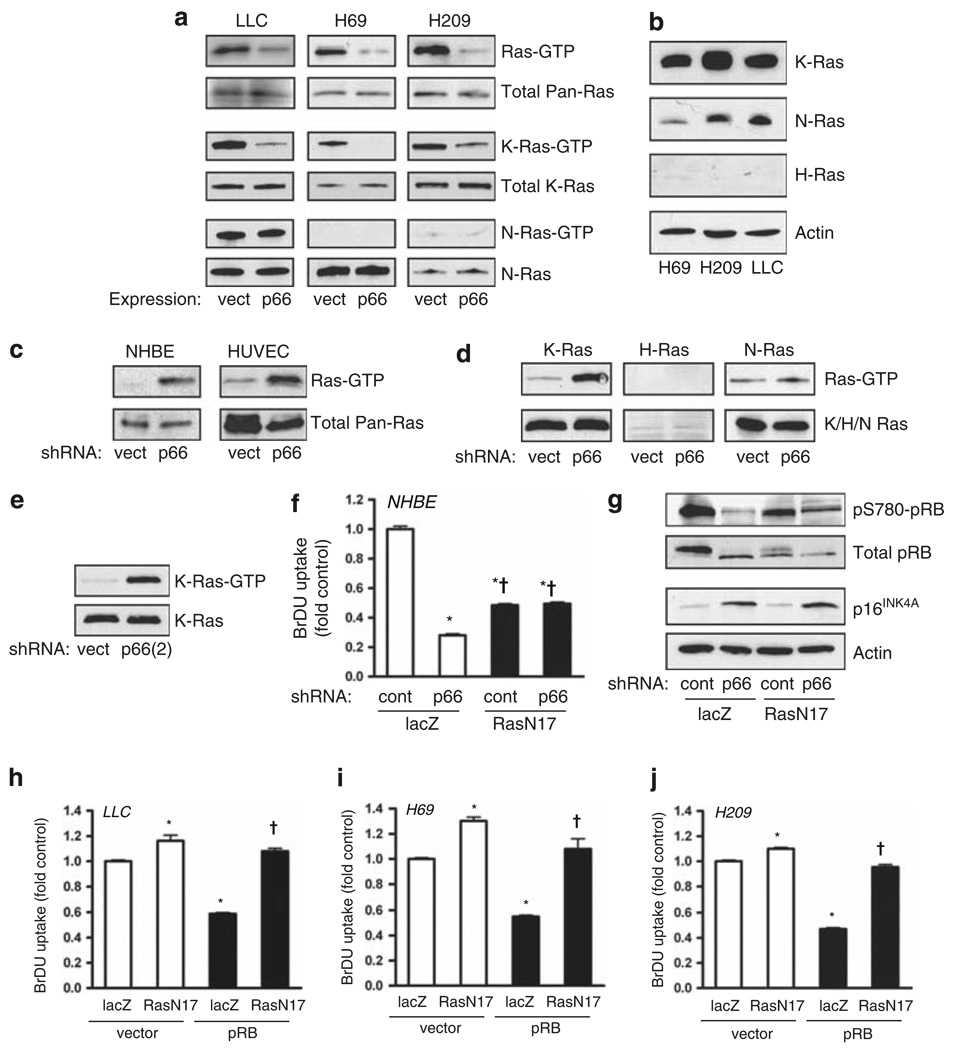
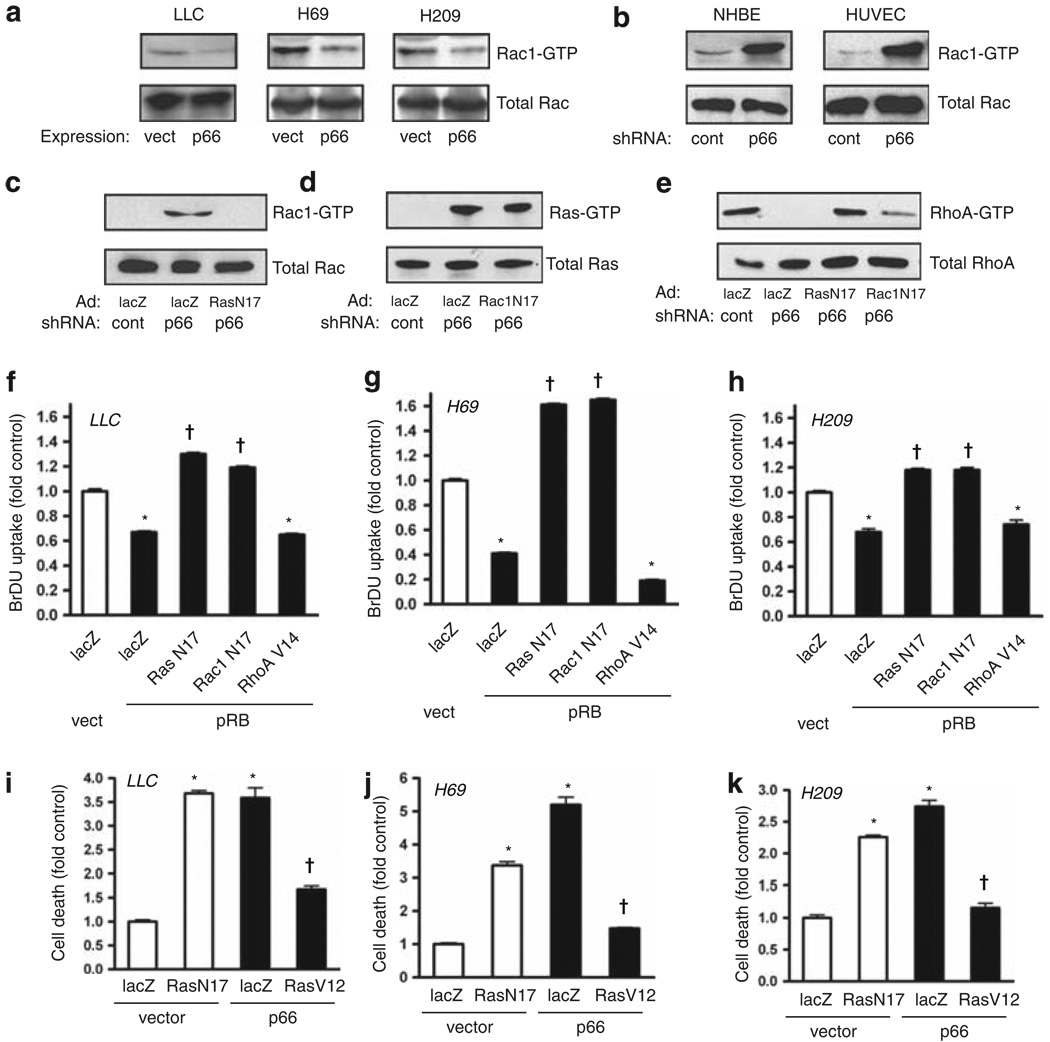
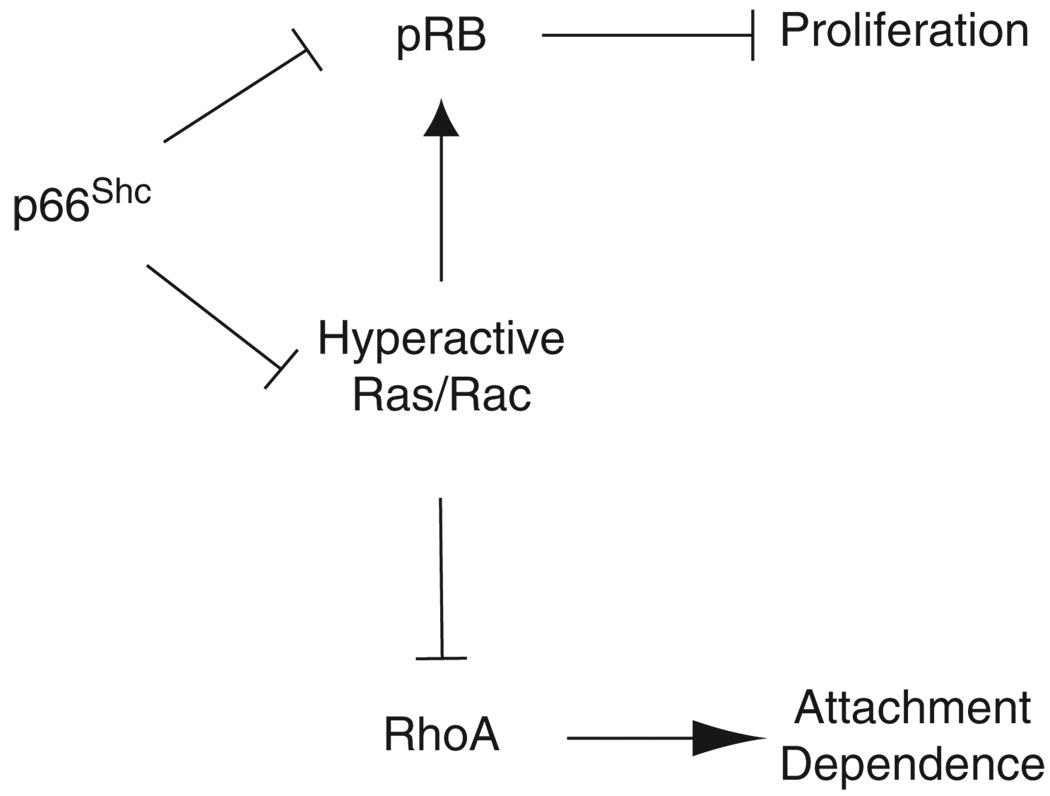
Normal tissue cells survive and proliferate only while anchored to solid substrate. Conversely, transformed cells both survive and proliferate following detachment, having lost attachment context through unclear mechanisms. p66(Shc) is a focal adhesion-associated protein that reports cell attachment through a RhoA-dependent mechanosensory test. We find that human small cell lung cancer (SCLC) cells and mouse Lewis lung carcinoma (LLC), which display aggressive metastatic behavior, lack both p66(Shc) and retinoblastoma (pRB) and bypass anoikis. Re-expression of p66(Shc) in these cells restores anoikis and provides striking protection from metastasis by LLC cells in vivo. Notably, knockdown of p66(Shc) in normal epithelial cells leads to unrestrained Ras activation, preventing anoikis through downstream suppression of RhoA but blocking proliferation in a pRB-dependent manner, thus mimicking oncogenic Ras. Conversely, LLC and SCLC cells display constitutive Ras activation necessary to bypass anoikis, which is reversed by re-expression of p66(Shc). p66(Shc) therefore coordinates Ras-dependent control of proliferation and anchorage sensation, which can be defeated in the evolution of highly metastatic tumors by combined loss of both p66(Shc) and pRB.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
p66(Shc) and Ras: controlling anoikis from the inside-out.Oncogene. 2010 Oct 14;29(41):5556-8. doi: 10.1038/onc.2010.347. Epub 2010 Aug 16. Oncogene. 2010. PMID: 20711240
Similar articles
-
p66(Shc) and Ras: controlling anoikis from the inside-out.Oncogene. 2010 Oct 14;29(41):5556-8. doi: 10.1038/onc.2010.347. Epub 2010 Aug 16. Oncogene. 2010. PMID: 20711240
-
Downregulated adaptor protein p66(Shc) mitigates autophagy process by low nutrient and enhances apoptotic resistance in human lung adenocarcinoma A549 cells.FEBS J. 2013 Sep;280(18):4522-30. doi: 10.1111/febs.12416. Epub 2013 Jul 25. FEBS J. 2013. PMID: 23815759
-
Detachment-Based Equilibrium of Anoikic Cell Death and Autophagic Cell Survival Through Adaptor Protein p66(Shc).Anat Rec (Hoboken). 2016 Mar;299(3):325-33. doi: 10.1002/ar.23299. Epub 2015 Dec 25. Anat Rec (Hoboken). 2016. PMID: 26643258
-
Shc and the mechanotransduction of cellular anchorage and metastasis.Small GTPases. 2019 Jan;10(1):64-71. doi: 10.1080/21541248.2016.1273172. Epub 2017 Feb 21. Small GTPases. 2019. PMID: 28632027 Free PMC article. Review.
-
p66(Shc) protein, oxidative stress, and cardiovascular complications of diabetes: the missing link.J Mol Med (Berl). 2009 Sep;87(9):885-91. doi: 10.1007/s00109-009-0499-3. Epub 2009 Jul 10. J Mol Med (Berl). 2009. PMID: 19590843 Review.
Cited by
-
Anoikis resistance of small airway epithelium is involved in the progression of chronic obstructive pulmonary disease.Front Immunol. 2023 Apr 5;14:1155478. doi: 10.3389/fimmu.2023.1155478. eCollection 2023. Front Immunol. 2023. PMID: 37090717 Free PMC article.
-
Ras, TrkB, and ShcA Protein Expression Patterns in Pediatric Brain Tumors.J Clin Med. 2021 May 20;10(10):2219. doi: 10.3390/jcm10102219. J Clin Med. 2021. PMID: 34065573 Free PMC article.
-
Mutant p53 disrupts role of ShcA protein in balancing Smad protein-dependent and -independent signaling activity of transforming growth factor-β (TGF-β).J Biol Chem. 2011 Dec 23;286(51):44023-44034. doi: 10.1074/jbc.M111.265397. Epub 2011 Oct 28. J Biol Chem. 2011. PMID: 22039050 Free PMC article.
-
Transcriptional coregualtor NUPR1 maintains tamoxifen resistance in breast cancer cells.Cell Death Dis. 2021 Feb 4;12(2):149. doi: 10.1038/s41419-021-03442-z. Cell Death Dis. 2021. PMID: 33542201 Free PMC article.
-
The G protein-coupled estrogen receptor-1, GPER-1, promotes fibrillogenesis via a Shc-dependent pathway resulting in anchorage-independent growth.Horm Cancer. 2014 Dec;5(6):390-404. doi: 10.1007/s12672-014-0195-9. Epub 2014 Aug 6. Horm Cancer. 2014. PMID: 25096985 Free PMC article.
References
-
- Attwell S, Roskelley C, Dedhar S. The integrin-linked kinase (ILK) suppresses anoikis. Oncogene. 2000;19:3811–3815. - PubMed
-
- Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430:1034–1039. - PubMed
-
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
