

Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy?
- PMID: 20676146
- PMCID: PMC3221965
- DOI: 10.1038/nrmicro2387
Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy?
Abstract
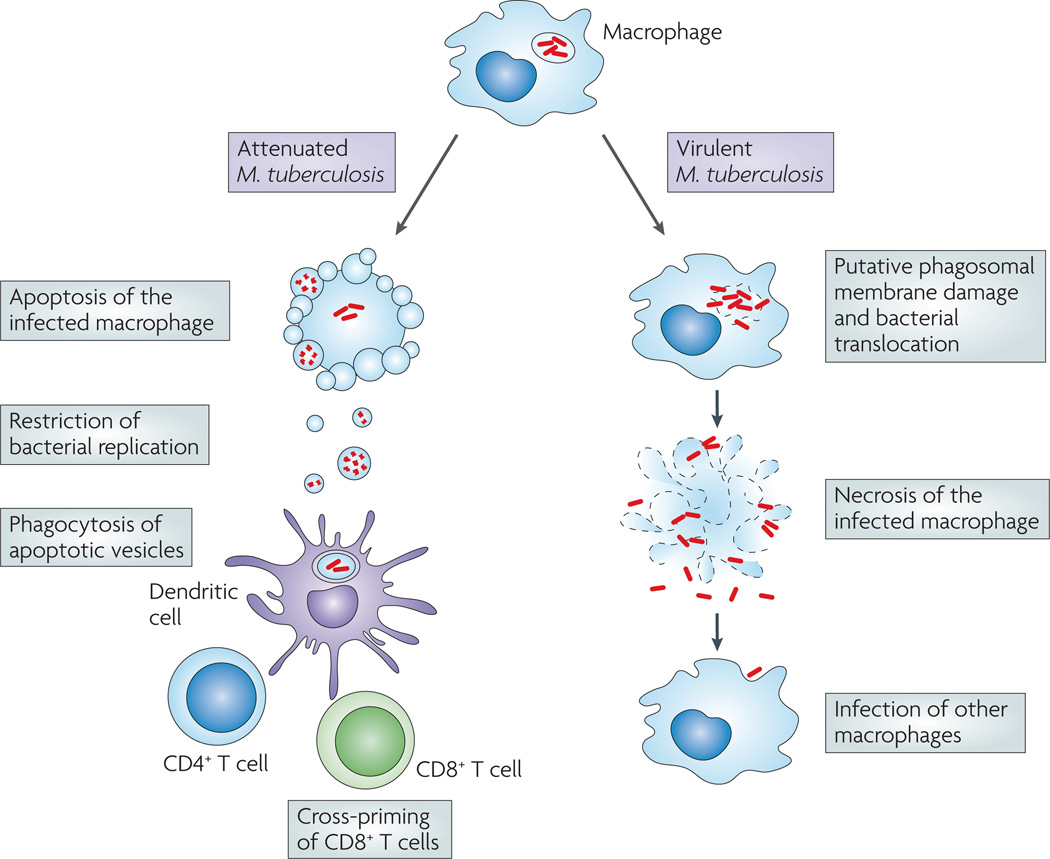
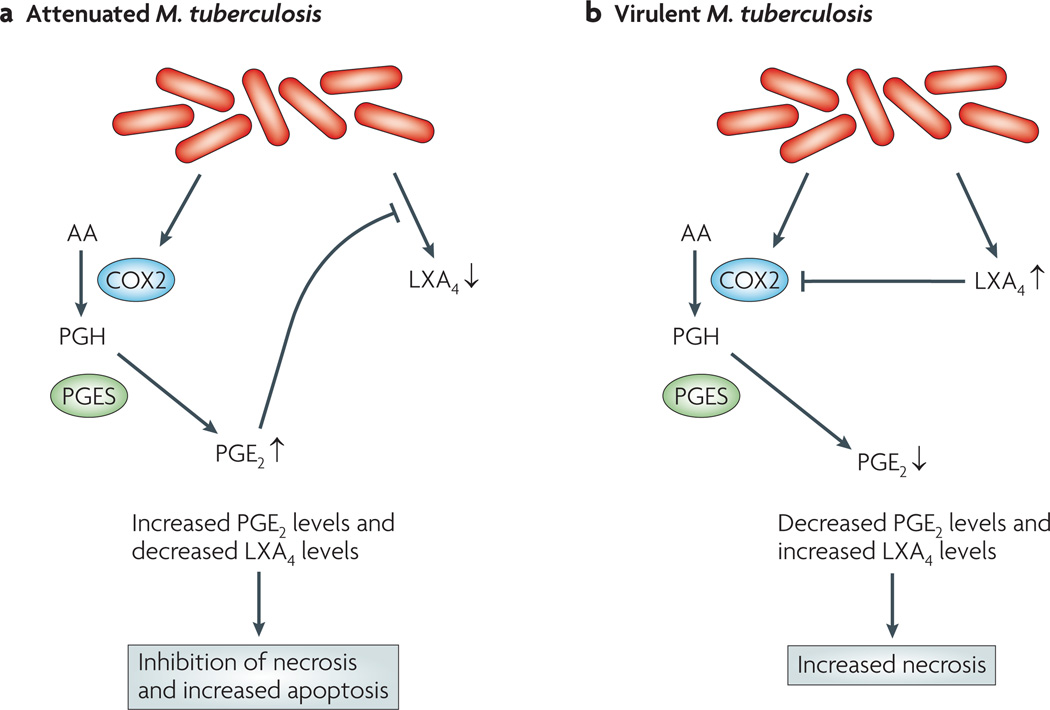
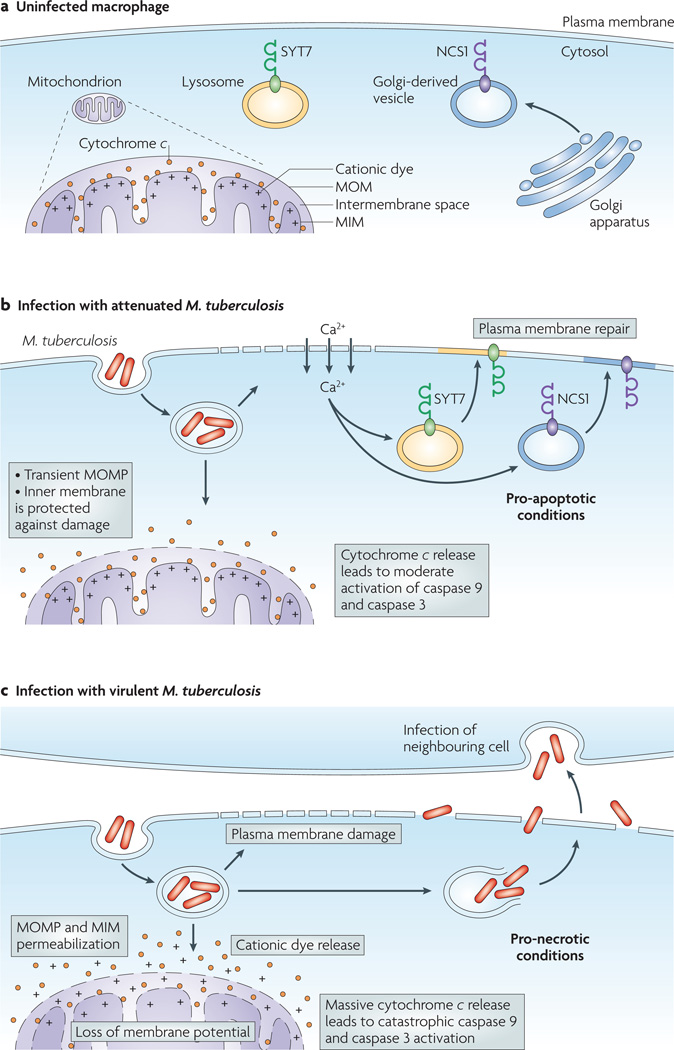
Virulent Mycobacterium tuberculosis inhibits apoptosis and triggers necrosis of host macrophages to evade innate immunity and delay the initiation of adaptive immunity. By contrast, attenuated M. tuberculosis induces macrophage apoptosis, an innate defence mechanism that reduces bacterial viability. In this Opinion article, we describe how virulent M. tuberculosis blocks production of the eicosanoid lipid mediator prostaglandin E(2) (PGE(2)). PGE(2) production by infected macrophages prevents mitochondrial damage and initiates plasma membrane repair, two processes that are crucial for preventing necrosis and inducing apoptosis. Thus, M. tuberculosis-mediated modulation of eicosanoid production determines the death modality of the infected macrophage, which in turn has a substantial impact on the outcome of infection.
Figures
References
-
- Bhatt K, Salgame P. Host innate immune response to Mycobacterium tuberculosis. J. Clin. Immunol. 2007;27:347–362. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
