

Integrative analysis of large scale expression profiles reveals core transcriptional response and coordination between multiple cellular processes in a cyanobacterium
- PMID: 20678200
- PMCID: PMC2924297
- DOI: 10.1186/1752-0509-4-105
Integrative analysis of large scale expression profiles reveals core transcriptional response and coordination between multiple cellular processes in a cyanobacterium
Abstract
Background: Cyanobacteria are the only known prokaryotes capable of oxygenic photosynthesis. They play significant roles in global biogeochemical cycles and carbon sequestration, and have recently been recognized as potential vehicles for production of renewable biofuels. Synechocystis sp. PCC 6803 has been extensively used as a model organism for cyanobacterial studies. DNA microarray studies in Synechocystis have shown varying degrees of transcriptome reprogramming under altered environmental conditions. However, it is not clear from published work how transcriptome reprogramming affects pre-existing networks of fine-tuned cellular processes.
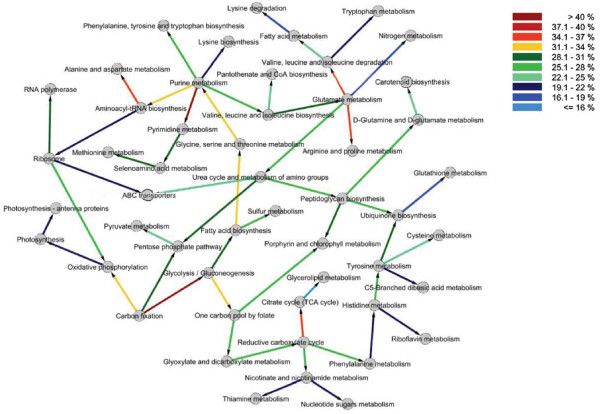
Results: We have integrated 163 transcriptome data sets generated in response to numerous environmental and genetic perturbations in Synechocystis. Our analyses show that a large number of genes, defined as the core transcriptional response (CTR), are commonly regulated under most perturbations. The CTR contains nearly 12% of Synechocystis genes found on its chromosome. The majority of genes in the CTR are involved in photosynthesis, translation, energy metabolism and stress protection. Our results indicate that a large number of differentially regulated genes identified in most reported studies in Synechocystis under different perturbations are associated with the general stress response. We also find that a majority of genes in the CTR are coregulated with 25 regulatory genes. Some of these regulatory genes have been implicated in cellular responses to oxidative stress, suggesting that reactive oxygen species are involved in the regulation of the CTR. A Bayesian network, based on the regulation of various KEGG pathways determined from the expression patterns of their associated genes, has revealed new insights into the coordination between different cellular processes.
Conclusion: We provide here the first integrative analysis of transcriptome data sets generated in a cyanobacterium. This compilation of data sets is a valuable resource to researchers for all cyanobacterial gene expression related queries. Importantly, our analysis provides a global description of transcriptional reprogramming under different perturbations and a basic framework to understand the strategies of cellular adaptations in Synechocystis.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
