

Manipulation of conformational change in proteins by single-residue perturbations
- PMID: 20682272
- PMCID: PMC2913187
- DOI: 10.1016/j.bpj.2010.05.020
Manipulation of conformational change in proteins by single-residue perturbations
Abstract
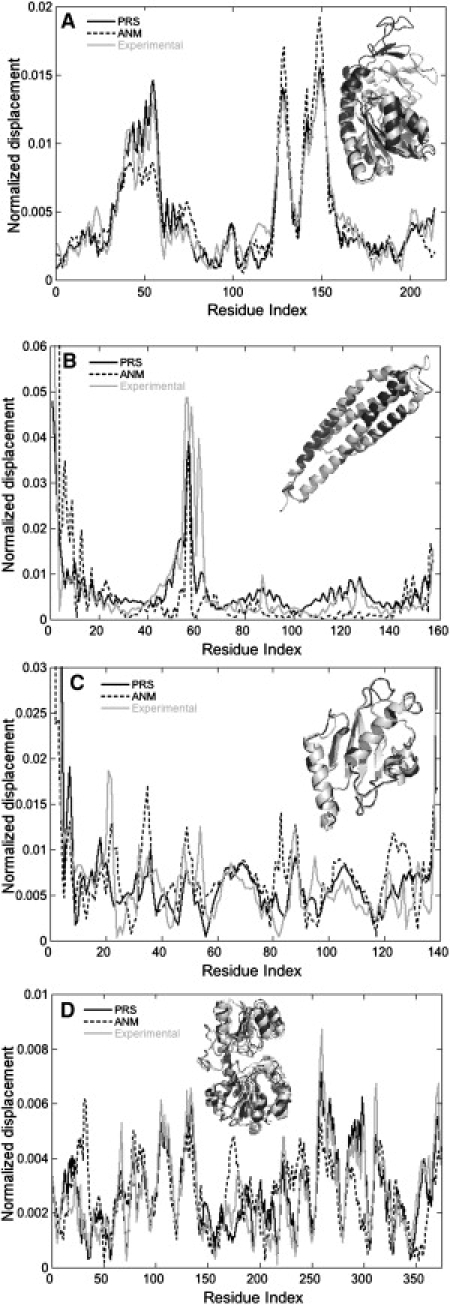
Using the perturbation-response scanning (PRS) technique, we study a set of 25 proteins that display a variety of conformational motions upon ligand binding (e.g., shear, hinge, allosteric). In most cases, PRS determines single residues that may be manipulated to achieve the resulting conformational change. PRS reveals that for some proteins, binding-induced conformational change may be achieved through the perturbation of residues scattered throughout the protein, whereas in others, perturbation of specific residues confined to a highly specific region is necessary. Overlaps between the experimental and PRS-calculated atomic displacement vectors are usually more descriptive of the conformational change than those obtained from a modal analysis of elastic network models. Furthermore, the largest overlaps obtained by the latter approach do not always appear in the most collective modes; there are cases where more than one mode yields comparable overlap sizes. We show that success of the modal analysis depends on an absence of redundant paths in the protein. PRS thus demonstrates that several relevant modes can be induced simultaneously by perturbing a single select residue on the protein. We also illustrate the biological relevance of applying PRS to the GroEL, adenylate kinase, myosin, and kinesin structures in detail by showing that the residues whose perturbation leads to precise conformational changes usually correspond to those experimentally determined to be functionally important.
2010 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures


Similar articles
-
A Coarse-Grained Methodology Identifies Intrinsic Mechanisms That Dissociate Interacting Protein Pairs.Front Mol Biosci. 2020 Aug 25;7:210. doi: 10.3389/fmolb.2020.00210. eCollection 2020. Front Mol Biosci. 2020. PMID: 33195399 Free PMC article.
-
Folding funnels and conformational transitions via hinge-bending motions.Cell Biochem Biophys. 1999;31(2):141-64. doi: 10.1007/BF02738169. Cell Biochem Biophys. 1999. PMID: 10593256 Review.
-
Can conformational change be described by only a few normal modes?Biophys J. 2006 Mar 1;90(5):1583-93. doi: 10.1529/biophysj.105.070045. Epub 2005 Dec 16. Biophys J. 2006. PMID: 16361336 Free PMC article.
-
Subtle pH differences trigger single residue motions for moderating conformations of calmodulin.J Chem Phys. 2011 Oct 21;135(15):155102. doi: 10.1063/1.3651807. J Chem Phys. 2011. PMID: 22029336
-
New structures of allosteric proteins revealing remarkable conformational changes.Curr Opin Struct Biol. 1996 Dec;6(6):824-9. doi: 10.1016/s0959-440x(96)80013-3. Curr Opin Struct Biol. 1996. PMID: 8994883 Review.
Cited by
-
Mutations in Antibody Fragments Modulate Allosteric Response Via Hydrogen-Bond Network Fluctuations.Biophys J. 2016 May 10;110(9):1933-42. doi: 10.1016/j.bpj.2016.03.033. Biophys J. 2016. PMID: 27166802 Free PMC article.
-
Surveying the Side-Chain Network Approach to Protein Structure and Dynamics: The SARS-CoV-2 Spike Protein as an Illustrative Case.Front Mol Biosci. 2020 Dec 18;7:596945. doi: 10.3389/fmolb.2020.596945. eCollection 2020. Front Mol Biosci. 2020. PMID: 33392257 Free PMC article. Review.
-
Substrate binding and channeling allosterically modulate the interactions within the AlkB-AlkG electron transfer complex.bioRxiv [Preprint]. 2025 Jun 27:2025.06.23.661152. doi: 10.1101/2025.06.23.661152. bioRxiv. 2025. PMID: 40667144 Free PMC article. Preprint.
-
Adaptive Evolution as a Predictor of Species-Specific Innate Immune Response.Mol Biol Evol. 2015 Jul;32(7):1717-29. doi: 10.1093/molbev/msv051. Epub 2015 Mar 10. Mol Biol Evol. 2015. PMID: 25758009 Free PMC article.
-
Fundamentals to function: Quantitative and scalable approaches for measuring protein stability.Cell Syst. 2021 Jun 16;12(6):547-560. doi: 10.1016/j.cels.2021.05.009. Cell Syst. 2021. PMID: 34139165 Free PMC article. Review.
References
-
- Case D.A. Normal-mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 1994;4:285–290.
-
- Hinsen K. Analysis of domain motions by approximate normal mode calculations. Proteins. 1998;33:417–429. - PubMed
-
- Tama F., Sanejouand Y.H. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001;14:1–6. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
