

Kinase-mediated quasi-dimers of EGFR
- PMID: 20682838
- PMCID: PMC2992368
- DOI: 10.1096/fj.10-166199
Kinase-mediated quasi-dimers of EGFR
Abstract
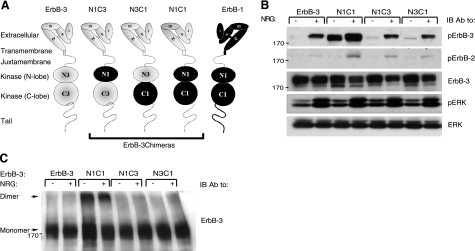
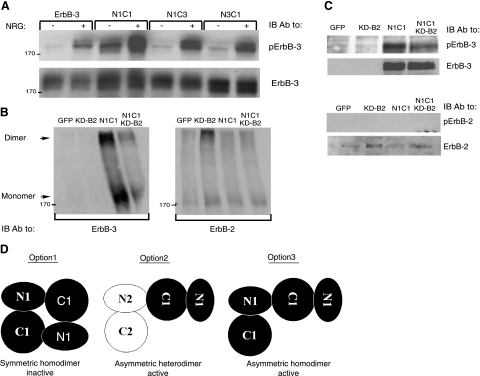
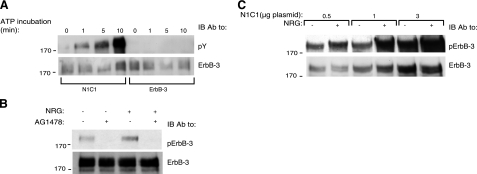
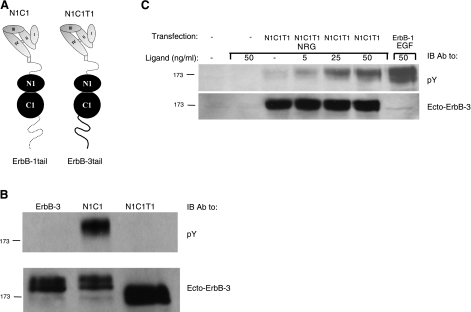
Ligand-induced dimerization of the epidermal growth factor receptor (ErbB-1/EGFR) involves conformational changes that expose an extracellular dimerization interface. Subsequent alterations within the cytoplasmic kinase domain, which culminate in tyrosine phosphorylation, are less understood. Our study addressed this question by using two strategies: a chimeric receptor approach employed ErbB-3, whose defective kinase domain was replaced by the respective part of EGFR. The implanted full-length kinase, unlike its subdomains, conferred dimerization and catalysis. The data infer that the kinase function of EGFR is restrained by the carboxyl tail; once grafted distally to the ectopic tail of ErbB-3, the kinase domain acquires quasi-dimerization and activation. In an attempt to alternatively refold the cytoplasmic tail, our other approach employed kinase inhibitors. Biophysical measurements and covalent cross-linking analyses showed that inhibitors targeting the active conformation of EGFR, in contrast to a compound recognizing the inactive conformation, induce quasi-dimers in a manner similar to the chimeric ErbB-3 molecule. Collectively, these observations unveil kinase domain-mediated quasi-dimers, which are regulated by an autoinhibitory carboxyl tail. On the basis of these observations, we propose that quasi-dimers precede formation of ligand-induced, fully active dimers, which are stabilized by both extracellular and intracellular receptor-receptor interactions.
Figures
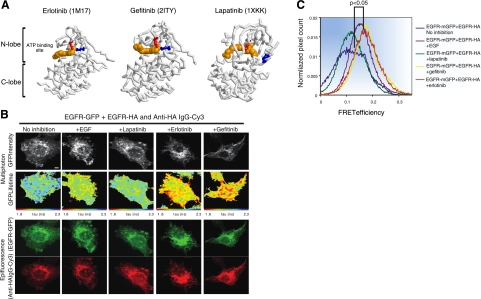
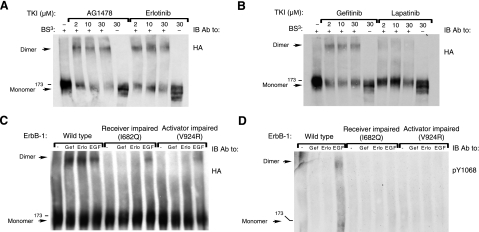
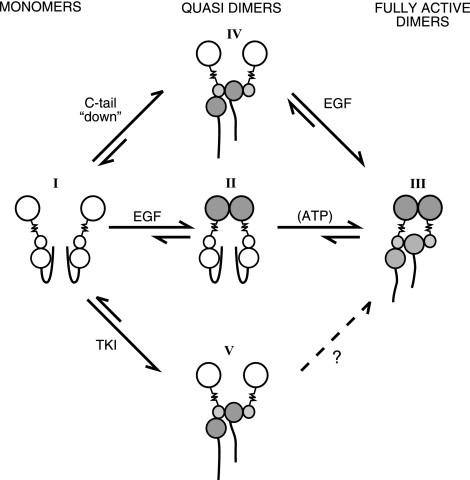
Similar articles
-
Mechanisms for kinase-mediated dimerization of the epidermal growth factor receptor.J Biol Chem. 2012 Nov 2;287(45):38244-53. doi: 10.1074/jbc.M112.414391. Epub 2012 Sep 17. J Biol Chem. 2012. PMID: 22988250 Free PMC article.
-
HER3 intracellular domains play a crucial role in HER3/HER2 dimerization and activation of downstream signaling pathways.Protein Cell. 2012 Oct;3(10):781-9. doi: 10.1007/s13238-012-2065-y. Epub 2012 Sep 15. Protein Cell. 2012. PMID: 22983903 Free PMC article.
-
Dynamic analysis of the epidermal growth factor (EGF) receptor-ErbB2-ErbB3 protein network by luciferase fragment complementation imaging.J Biol Chem. 2013 Oct 18;288(42):30773-30784. doi: 10.1074/jbc.M113.489534. Epub 2013 Sep 6. J Biol Chem. 2013. PMID: 24014028 Free PMC article.
-
Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers.Pharmacol Res. 2019 Jan;139:395-411. doi: 10.1016/j.phrs.2018.11.014. Epub 2018 Nov 27. Pharmacol Res. 2019. PMID: 30500458 Review.
-
Emerging Allosteric Mechanism of EGFR Activation in Physiological and Pathological Contexts.Biophys J. 2019 Jul 9;117(1):5-13. doi: 10.1016/j.bpj.2019.05.021. Epub 2019 May 28. Biophys J. 2019. PMID: 31202480 Free PMC article. Review.
Cited by
-
Lung injury and lung cancer caused by cigarette smoke-induced oxidative stress: Molecular mechanisms and therapeutic opportunities involving the ceramide-generating machinery and epidermal growth factor receptor.Antioxid Redox Signal. 2014 Nov 20;21(15):2149-74. doi: 10.1089/ars.2013.5469. Epub 2014 Jul 1. Antioxid Redox Signal. 2014. PMID: 24684526 Free PMC article. Review.
-
Effect of phosphorylation on EGFR dimer stability probed by single-molecule dynamics and FRET/FLIM.Biophys J. 2015 Mar 10;108(5):1013-26. doi: 10.1016/j.bpj.2015.01.005. Biophys J. 2015. PMID: 25762314 Free PMC article.
-
A point mutation in the extracellular domain of KIT promotes tumorigenesis of mast cells via ligand-independent auto-dimerization.Sci Rep. 2015 May 12;5:9775. doi: 10.1038/srep09775. Sci Rep. 2015. PMID: 25965812 Free PMC article.
-
Fluorescence Imaging of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Resistance in Non-Small Cell Lung Cancer.Cancers (Basel). 2022 Jan 28;14(3):686. doi: 10.3390/cancers14030686. Cancers (Basel). 2022. PMID: 35158954 Free PMC article. Review.
-
EGF receptor exposed to oxidative stress acquires abnormal phosphorylation and aberrant activated conformation that impairs canonical dimerization.PLoS One. 2011;6(8):e23240. doi: 10.1371/journal.pone.0023240. Epub 2011 Aug 10. PLoS One. 2011. PMID: 21853092 Free PMC article.
References
-
- Yarden Y., Sliwkowski M. X. (2001) Untangling the ErbB signalling network. Nat. Rev. Mol. Cell. Biol. 2, 127–137 - PubMed
-
- Garrett T. P., McKern N. M., Lou M., Elleman T. C., Adams T. E., Lovrecz G. O., Zhu H. J., Walker F., Frenkel M. J., Hoyne P. A., Jorissen R. N., Nice E. C., Burgess A. W., Ward C. W. (2002) Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 110, 763–773 - PubMed
-
- Ogiso H., Ishitani R., Nureki O., Fukai S., Yamanaka M., Kim J. H., Saito K., Sakamoto A., Inoue M., Shirouzu M., Yokoyama S. (2002) Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 110, 775–787 - PubMed
-
- Hynes N. E., MacDonald G. (2009) ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 21, 177–184 - PubMed
-
- Bublil E. M., Yarden Y. (2007) The EGF receptor family: spearheading a merger of signaling and therapeutics. Curr. Opin. Cell Biol. 19, 124–134 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
