

The significance, development and progress of high-throughput combinatorial histone code analysis
- PMID: 20683756
- PMCID: PMC11115713
- DOI: 10.1007/s00018-010-0475-7
The significance, development and progress of high-throughput combinatorial histone code analysis
Abstract
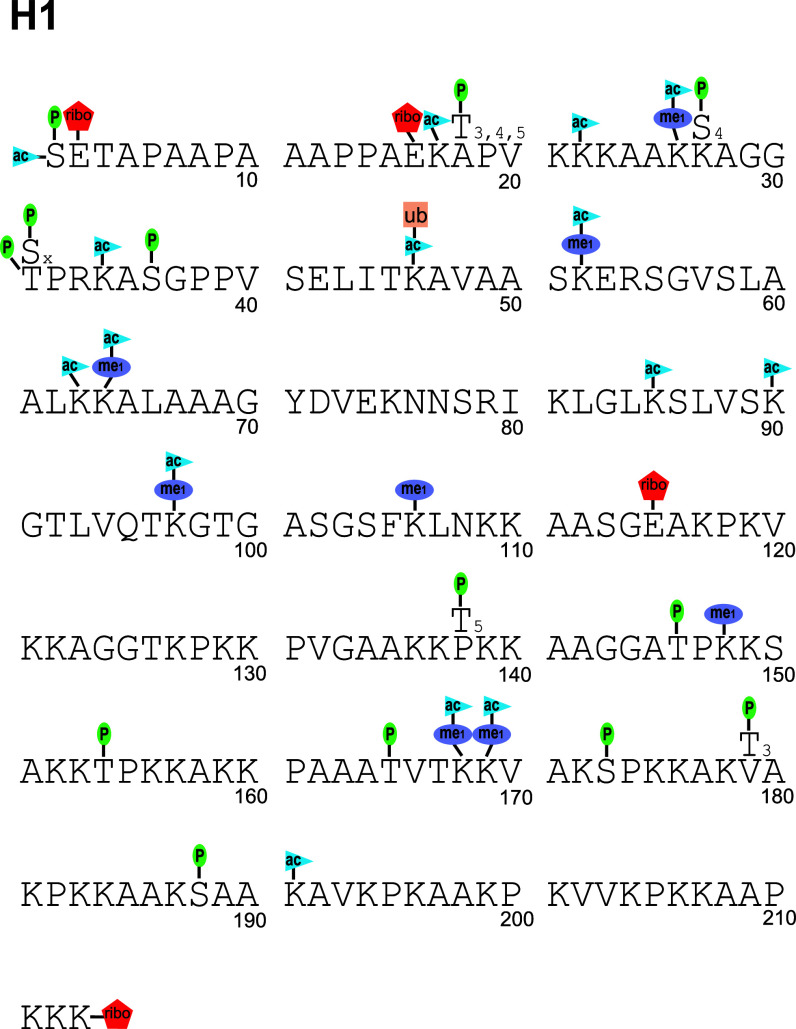
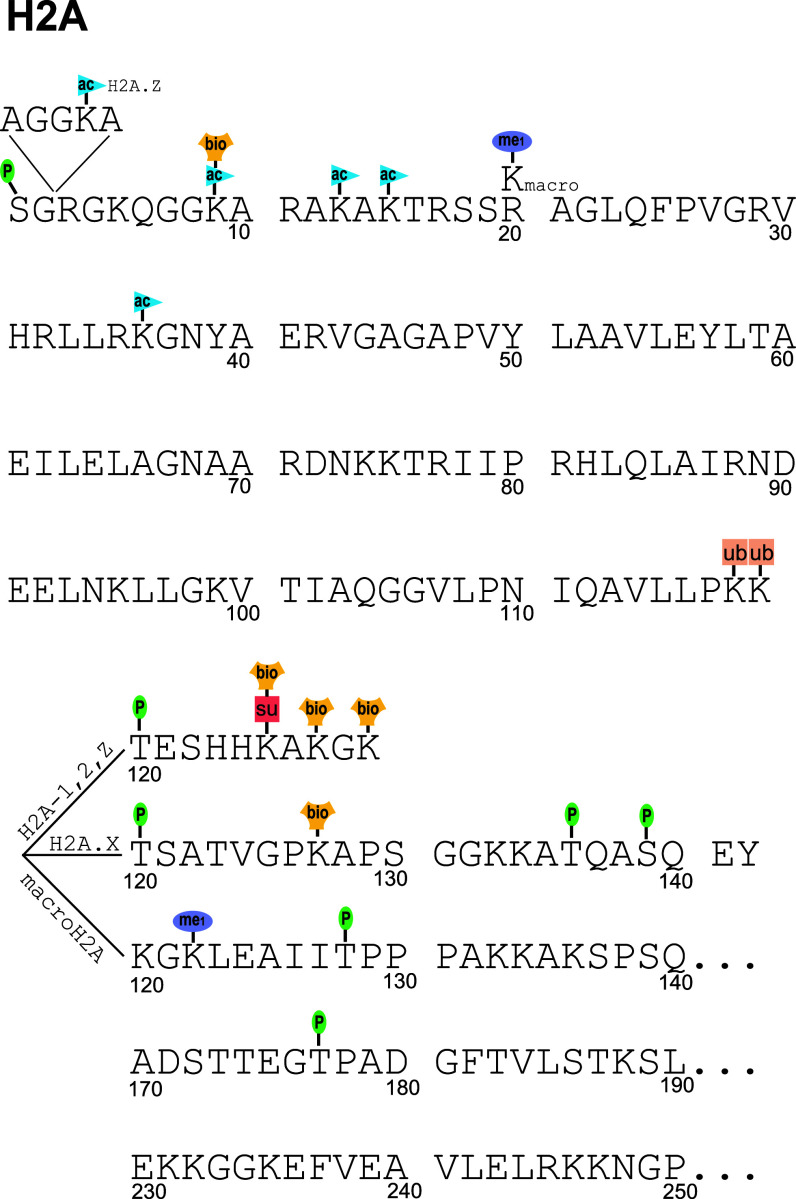
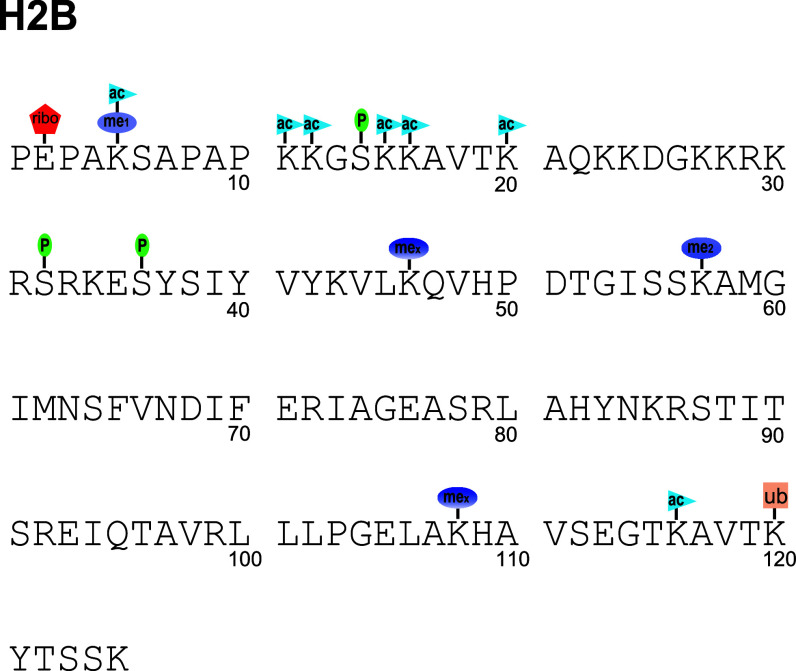
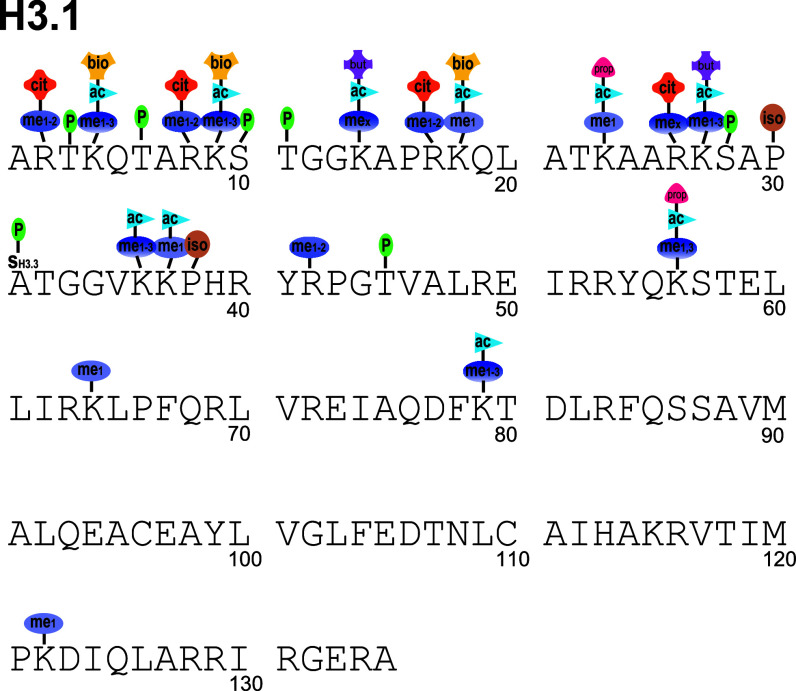
The physiological state of eukaryotic DNA is chromatin. Nucleosomes, which consist of DNA in complex with histones, are the fundamental unit of chromatin. The post-translational modifications (PTMs) of histones play a critical role in the control of gene transcription, epigenetics and other DNA-templated processes. It has been known for several years that these PTMs function in concert to allow for the storage and transduction of highly specific signals through combinations of modifications. This code, the combinatorial histone code, functions much like a bar code or combination lock providing the potential for massive information content. The capacity to directly measure these combinatorial histone codes has mostly been laborious and challenging, thus limiting efforts often to one or two samples. Recently, progress has been made in determining such information quickly, quantitatively and sensitively. Here we review both the historical and recent progress toward routine and rapid combinatorial histone code analysis.
Figures
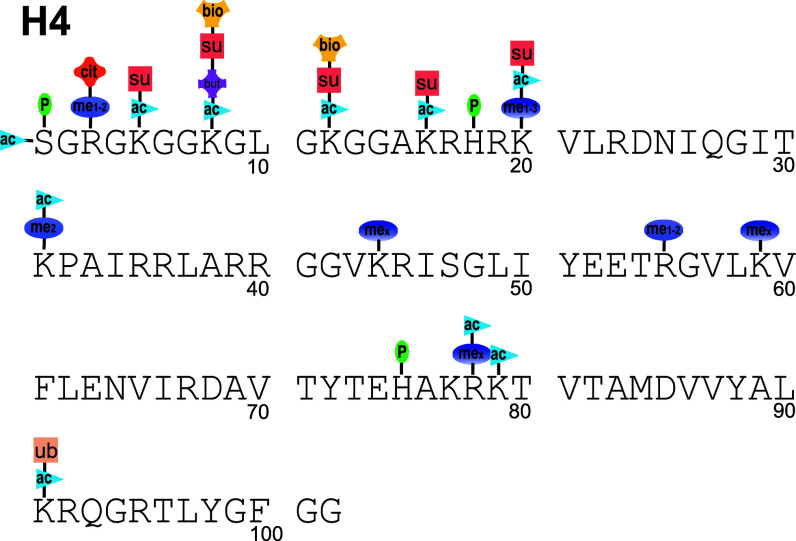
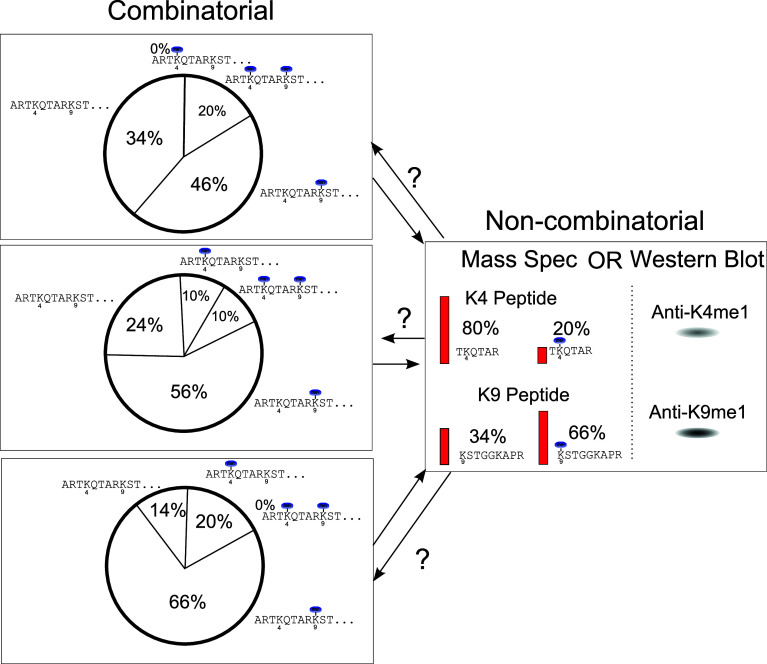
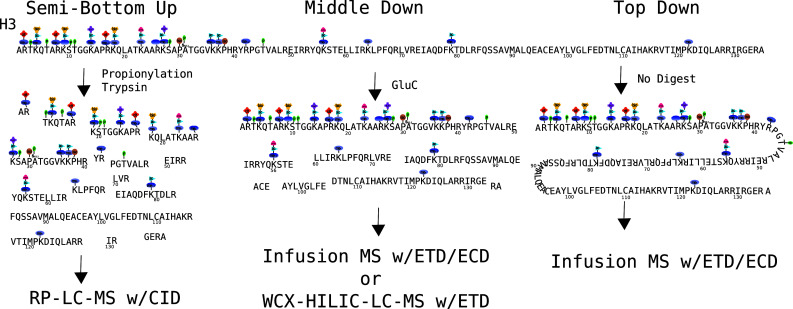
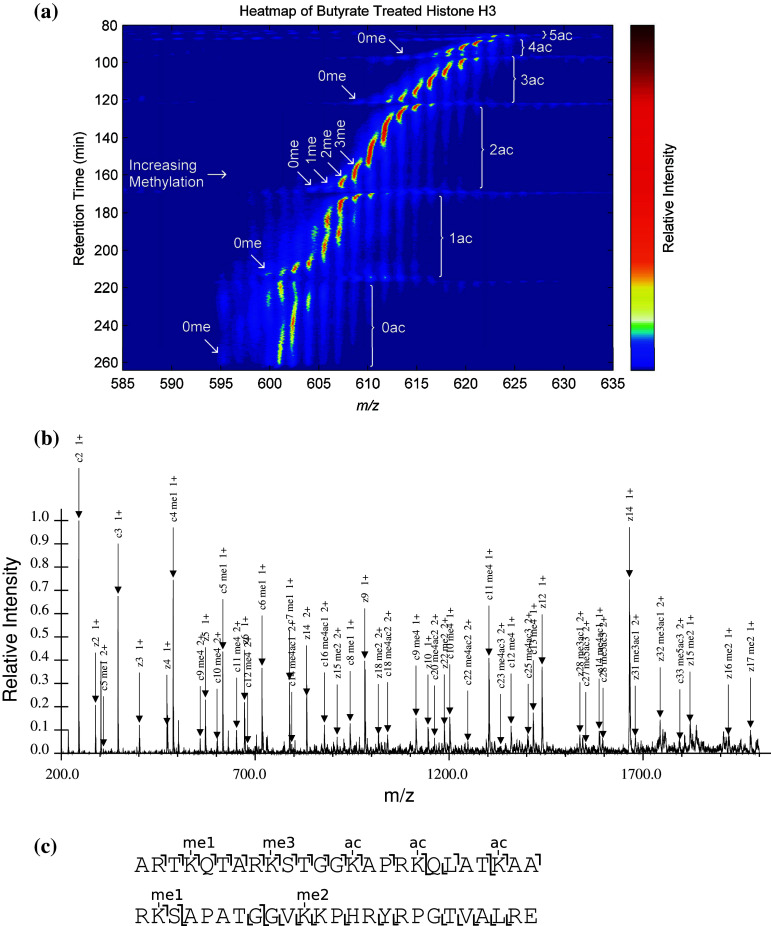
References
-
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. - PubMed
-
- Prohaska SJ, Stadler PF, Krakauer DC. Innovation in gene regulation: the case of chromatin computation. J Theor Biol. 2010;265(1):27–44. - PubMed
-
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. - PubMed
-
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. - PubMed
-
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
