

Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes
- PMID: 20683928
- PMCID: PMC3048164
- DOI: 10.1002/humu.21336
Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes
Abstract
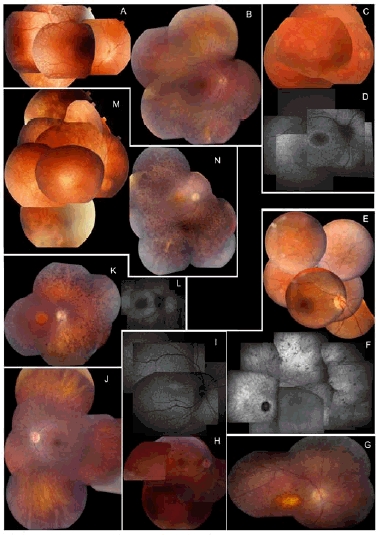
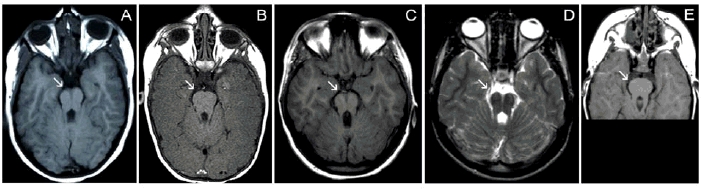
Leber Congenital Amaurosis (LCA), the most severe inherited retinal dystrophy, is genetically heterogeneous, with 14 genes accounting for 70% of patients. Here, 91 LCA probands underwent LCA chip analysis and subsequent sequencing of 6 genes (CEP290, CRB1, RPE65, GUCY2D, AIPL1and CRX), revealing mutations in 69% of the cohort, with major involvement of CEP290 (30%). In addition, 11 patients with early-onset retinal dystrophy (EORD) and 13 patients with Senior-Loken syndrome (SLS), LCA-Joubert syndrome (LCA-JS) or cerebello-oculo-renal syndrome (CORS) were included. Exhaustive re-inspection of the overall phenotypes in our LCA cohort revealed novel insights mainly regarding the CEP290-related phenotype. The AHI1 gene was screened as a candidate modifier gene in three patients with the same CEP290 genotype but different neurological involvement. Interestingly, a heterozygous novel AHI1 mutation, p.Asn811Lys, was found in the most severely affected patient. Moreover, AHI1 screening in five other patients with CEP290-related disease and neurological involvement revealed a second novel missense variant, p.His758Pro, in one LCA patient with mild mental retardation and autism. These two AHI1 mutations might thus represent neurological modifiers of CEP290-related disease.
© 2010 Wiley-Liss, Inc.
Figures
Similar articles
-
Differential macular morphology in patients with RPE65-, CEP290-, GUCY2D-, and AIPL1-related Leber congenital amaurosis.Invest Ophthalmol Vis Sci. 2010 May;51(5):2608-14. doi: 10.1167/iovs.09-3734. Epub 2009 Dec 3. Invest Ophthalmol Vis Sci. 2010. PMID: 19959640 Free PMC article.
-
Clinical and molecular findings in a cohort of 152 Brazilian severe early onset inherited retinal dystrophy patients.Am J Med Genet C Semin Med Genet. 2020 Sep;184(3):728-752. doi: 10.1002/ajmg.c.31828. Epub 2020 Aug 31. Am J Med Genet C Semin Med Genet. 2020. PMID: 32865313
-
Genetic and clinical findings in a Chinese cohort with Leber congenital amaurosis and early onset severe retinal dystrophy.Br J Ophthalmol. 2020 Jul;104(7):932-937. doi: 10.1136/bjophthalmol-2019-314281. Epub 2019 Oct 19. Br J Ophthalmol. 2020. PMID: 31630094
-
Leber's Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations.Genes (Basel). 2021 Aug 19;12(8):1261. doi: 10.3390/genes12081261. Genes (Basel). 2021. PMID: 34440435 Free PMC article. Review.
-
Leber congenital amaurosis: genes, proteins and disease mechanisms.Prog Retin Eye Res. 2008 Jul;27(4):391-419. doi: 10.1016/j.preteyeres.2008.05.003. Epub 2008 Jun 1. Prog Retin Eye Res. 2008. PMID: 18632300 Review.
Cited by
-
Species-dependent splice recognition of a cryptic exon resulting from a recurrent intronic CEP290 mutation that causes congenital blindness.Int J Mol Sci. 2015 Mar 9;16(3):5285-98. doi: 10.3390/ijms16035285. Int J Mol Sci. 2015. PMID: 25761237 Free PMC article.
-
Molecular and clinical analysis of 27 German patients with Leber congenital amaurosis.PLoS One. 2018 Dec 21;13(12):e0205380. doi: 10.1371/journal.pone.0205380. eCollection 2018. PLoS One. 2018. PMID: 30576320 Free PMC article. Clinical Trial.
-
Ciliary genes arl13b, ahi1 and cc2d2a differentially modify expression of visual acuity phenotypes but do not enhance retinal degeneration due to mutation of cep290 in zebrafish.PLoS One. 2019 Apr 10;14(4):e0213960. doi: 10.1371/journal.pone.0213960. eCollection 2019. PLoS One. 2019. PMID: 30970040 Free PMC article.
-
Genotypic and phenotypic characterization of the Sdccag8Tn(sb-Tyr)2161B.CA1C2Ove mouse model.PLoS One. 2018 Feb 14;13(2):e0192755. doi: 10.1371/journal.pone.0192755. eCollection 2018. PLoS One. 2018. PMID: 29444170 Free PMC article.
-
Comprehensive genotyping reveals RPE65 as the most frequently mutated gene in Leber congenital amaurosis in Denmark.Eur J Hum Genet. 2016 Jul;24(7):1071-9. doi: 10.1038/ejhg.2015.241. Epub 2015 Dec 2. Eur J Hum Genet. 2016. PMID: 26626312 Free PMC article.
References
-
- Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, Esculpavit C, Toutain A, Moraine C, Parent P, Marcorelles P, Dauge MC, Roume J, Le Merrer M, Meiner V, Meir K, Menez F, Beaufrere AM, Francannet C, Tantau J, Sinico M, Dumez Y, MacDonald F, Munnich A, Lyonnet S, Gubler MC, Genin E, Johnson CA, Vekemans M, Encha-Razavi F, Attie-Bitach T. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet. 2007;81(1):170–9. - PMC - PubMed
-
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008;358(21):2231–9. - PubMed
-
- Bowne SJ, Sullivan LS, Mortimer SE, Hedstrom L, Zhu J, Spellicy CJ, Gire AI, Hughbanks-Wheaton D, Birch DG, Lewis RA, Heckenlively JR, Daiger SP. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2006;47(1):34–42. - PMC - PubMed
-
- Brancati F, Barrano G, Silhavy JL, Marsh SE, Travaglini L, Bielas SL, Amorini M, Zablocka D, Kayserili H, Al-Gazali L, Bertini E, Boltshauser E, D'Hooghe M, Fazzi E, Fenerci EY, Hennekam RC, Kiss A, Lees MM, Marco E, Phadke SR, Rigoli L, Romano S, Salpietro CD, Sherr EH, Signorini S, Stromme P, Stuart B, Sztriha L, Viskochil DH, Yuksel A, Dallapiccola B, Valente EM, Gleeson JG. CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am J Hum Genet. 2007;81(1):104–13. - PMC - PubMed
-
- Chang B, Khanna H, Hawes N, Jimeno D, He S, Lillo C, Parapuram SK, Cheng H, Scott A, Hurd RE, Sayer JA, Otto EA, Attanasio M, O'Toole JF, Jin G, Shou C, Hildebrandt F, Williams DS, Heckenlively JR, Swaroop A. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd1 6 mouse. Hum Mol Genet. 2006;15(11):1847–57. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
