

RNA structure determination using SAXS data
- PMID: 20684627
- PMCID: PMC3164809
- DOI: 10.1021/jp1057308
RNA structure determination using SAXS data
Abstract
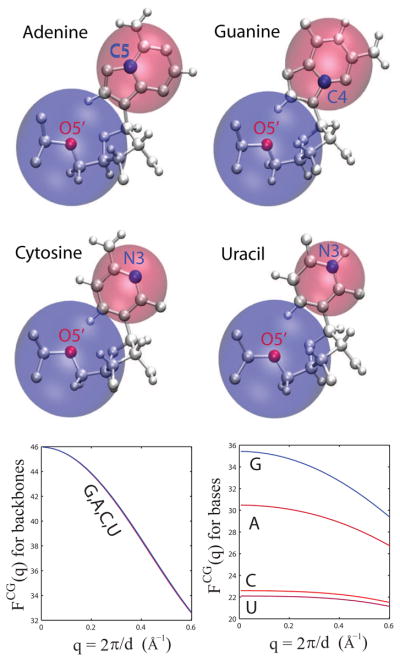
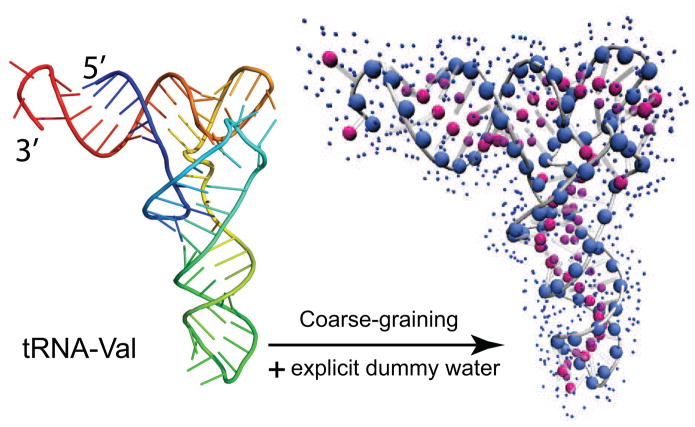
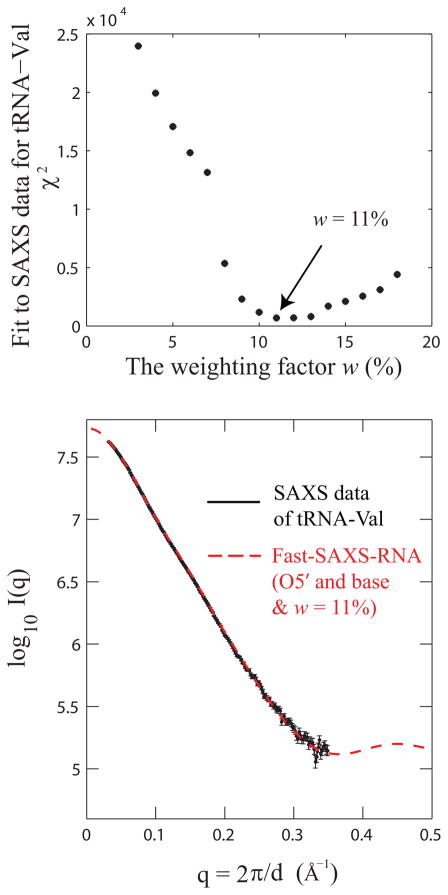
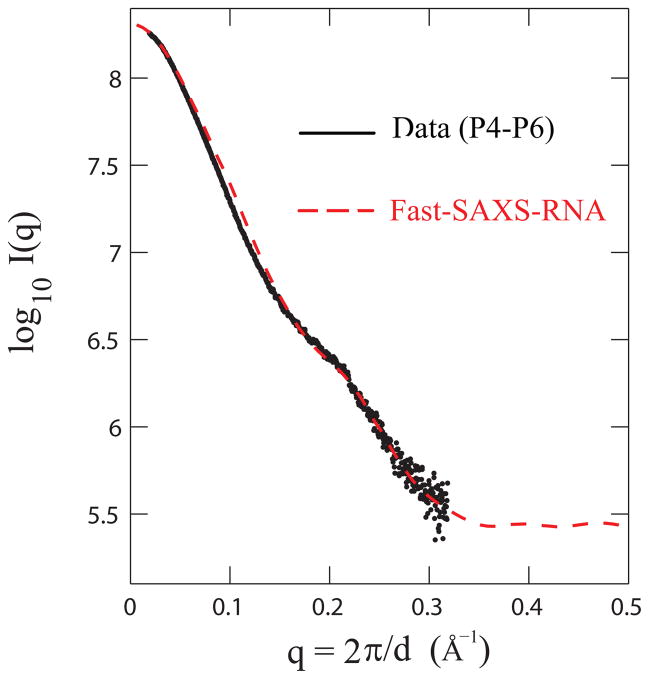
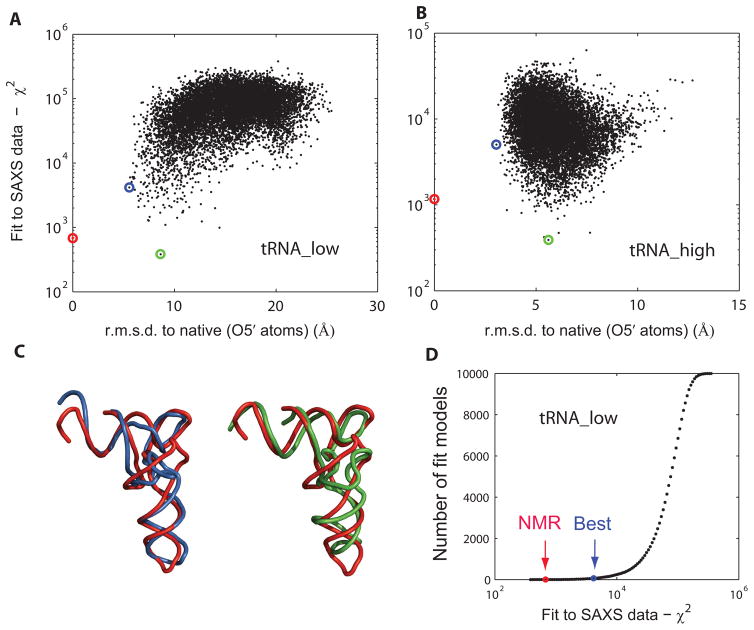
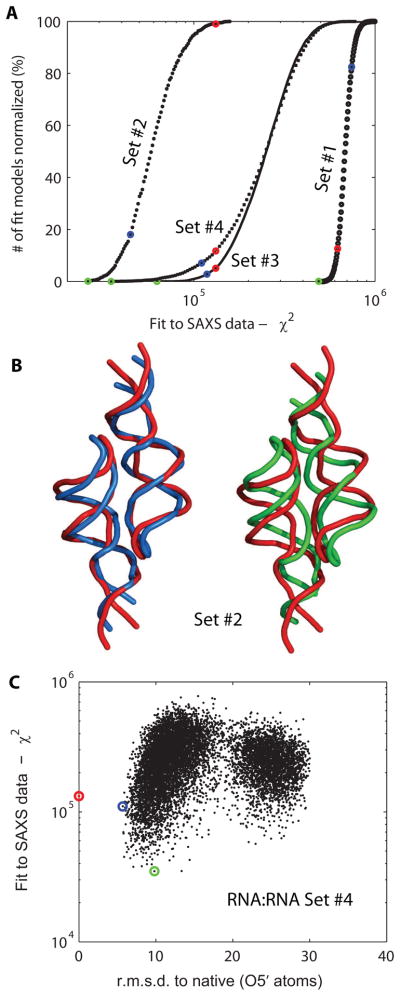
Exploiting the experimental information from small-angle X-ray solution scattering (SAXS) in conjunction with structure prediction algorithms can be advantageous in the case of ribonucleic acids (RNA), where global restraints on the 3D fold are often lacking. Traditional usage of SAXS data often starts by attempting to reconstruct the molecular shape ab initio, which is subsequently used to assess the quality of a model. Here, an alternative strategy is explored whereby the models from a very large decoy set are directly sorted according to their fit to the SAXS data. For rapid computation of SAXS patterns, the method developed here makes use of a coarse-grained representation of RNA. It also accounts for the explicit treatment of the contribution to the scattering of water molecules and ions surrounding the RNA. The method, called Fast-SAXS-RNA, is first calibrated using a tRNA (tRNA-val) and then tested on the P4-P6 fragment of group I intron (P4-P6). Fast-SAXS-RNA is then used as a filter for decoy models generated by the MC-Fold and MC-Sym pipeline, a suite of RNA 3D all-atom structure algorithms that encode and exploit RNA 3D architectural principles. The ability of Fast-SAXS-RNA to discriminate native folds is tested against three widely used RNA molecules in molecular modeling benchmarks: the tRNA, the P4-P6, and a synthetic hairpin suspected to assemble into a homodimer. For each molecule, a large pool of decoys are generated, scored, and ranked using Fast-SAXS-RNA. The method is able to identify low-rmsd models among top ranking structures, for both tRNA and P4-P6. For the hairpin, the approach correctly identifies the dimeric state as the solution structure over the monomeric state and alternative secondary structures. The method offers a powerful strategy for recognizing native RNA conformations as well as multimeric assemblies and alternative secondary structures, thus enabling high-throughput RNA structure determination using SAXS data.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
