

Nonparametric identification of regulatory interactions from spatial and temporal gene expression data
- PMID: 20684787
- PMCID: PMC2933715
- DOI: 10.1186/1471-2105-11-413
Nonparametric identification of regulatory interactions from spatial and temporal gene expression data
Abstract
Background: The correlation between the expression levels of transcription factors and their target genes can be used to infer interactions within animal regulatory networks, but current methods are limited in their ability to make correct predictions.
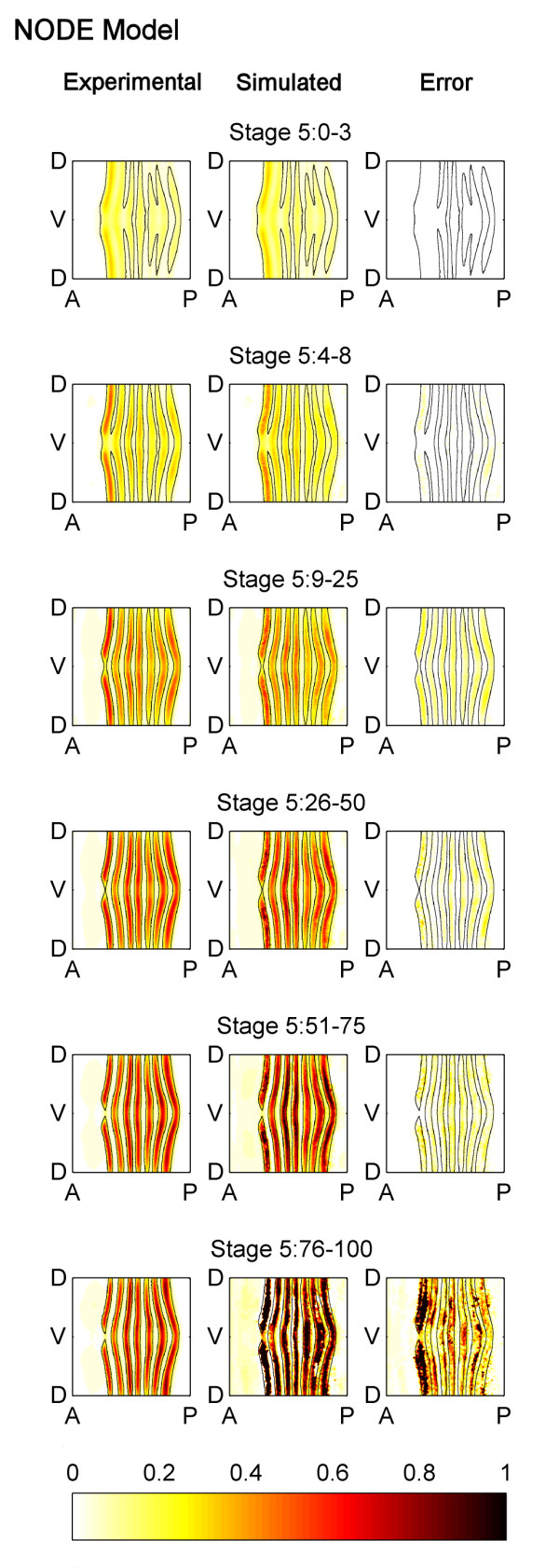
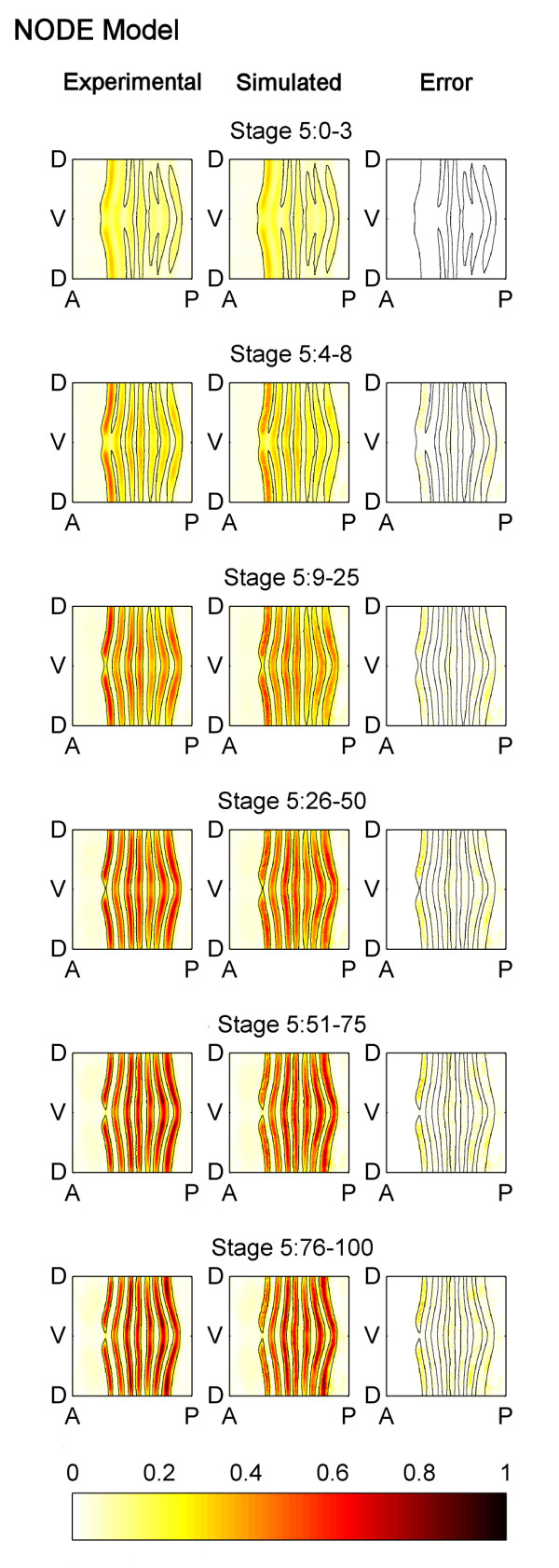
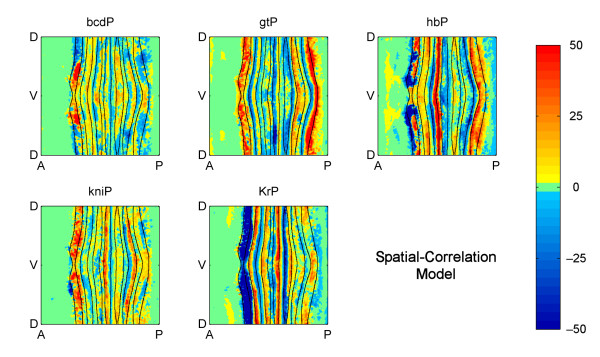
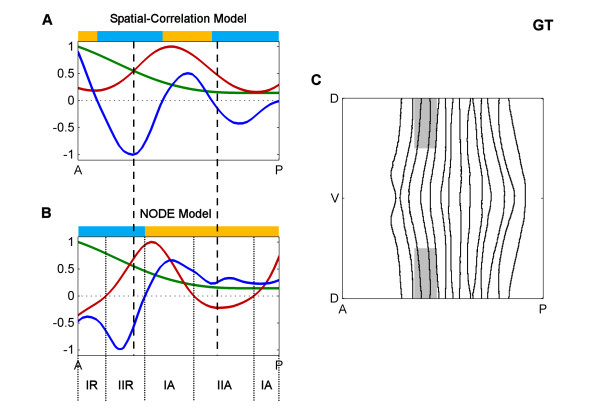
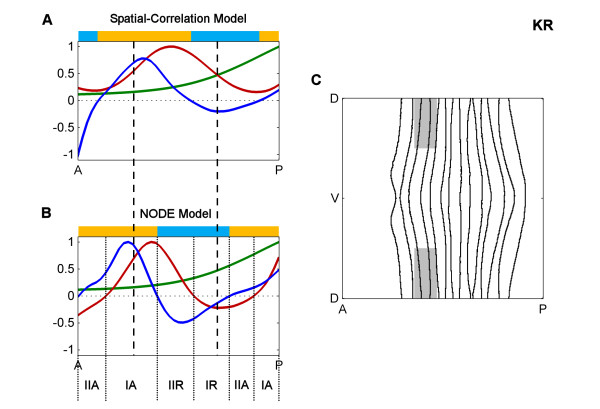
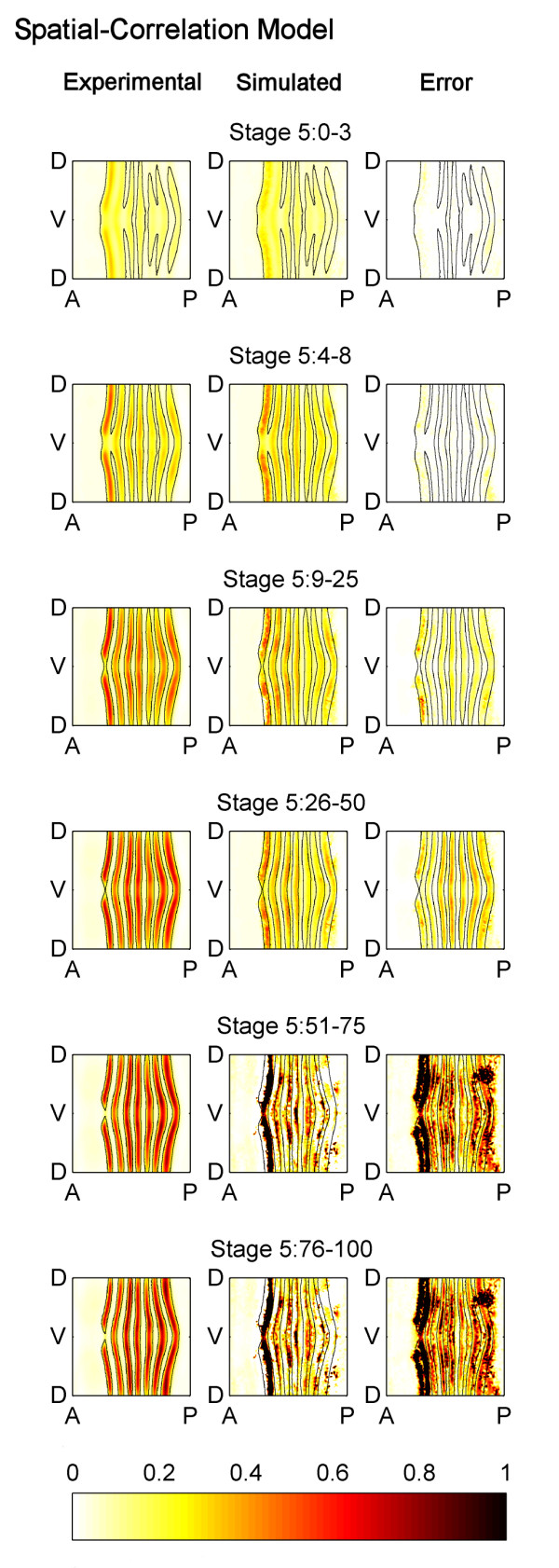
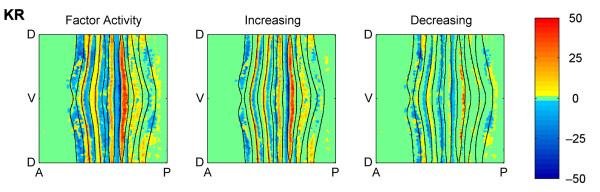
Results: Here we describe a novel approach which uses nonparametric statistics to generate ordinary differential equation (ODE) models from expression data. Compared to other dynamical methods, our approach requires minimal information about the mathematical structure of the ODE; it does not use qualitative descriptions of interactions within the network; and it employs new statistics to protect against over-fitting. It generates spatio-temporal maps of factor activity, highlighting the times and spatial locations at which different regulators might affect target gene expression levels. We identify an ODE model for eve mRNA pattern formation in the Drosophila melanogaster blastoderm and show that this reproduces the experimental patterns well. Compared to a non-dynamic, spatial-correlation model, our ODE gives 59% better agreement to the experimentally measured pattern. Our model suggests that protein factors frequently have the potential to behave as both an activator and inhibitor for the same cis-regulatory module depending on the factors' concentration, and implies different modes of activation and repression.
Conclusions: Our method provides an objective quantification of the regulatory potential of transcription factors in a network, is suitable for both low- and moderate-dimensional gene expression datasets, and includes improvements over existing dynamic and static models.
Figures




References
-
- Markowetz F, Spang R. Inferring cellular networks - a review. BMC Bioinformatics. 2007;8(Suppl 6):S5. doi: 10.1186/1471-2105-8-S6-S5. http://www.biomedcentral.com/1471-2105/8/S6/S5 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
