

Sodium channel molecular conformations and antiarrhythmic drug affinity
- PMID: 20685573
- PMCID: PMC2917343
- DOI: 10.1016/j.tcm.2010.03.002
Sodium channel molecular conformations and antiarrhythmic drug affinity
Abstract
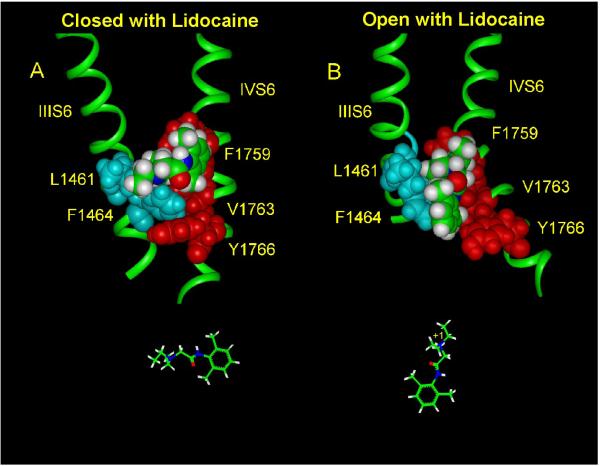
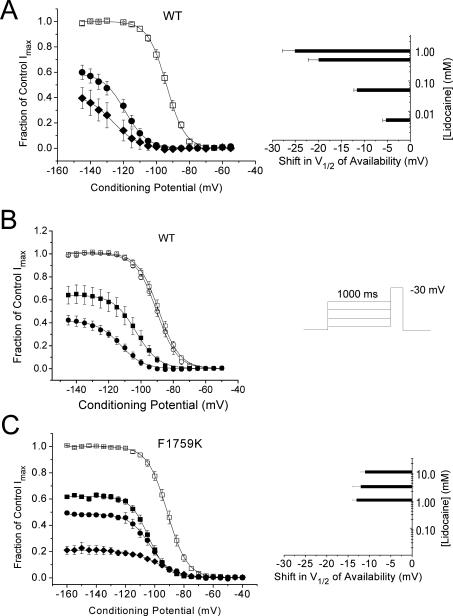
Class I cardiac antiarrhythmic drugs, for example, lidocaine, mexiletine, flecainide, quinidine, and procainamide, continue to play an important role in the therapy for cardiac arrhythmias because of the presence of use-dependent block. Lidocaine, as well as related drugs such as mepivacaine, bupivacaine, and cocaine, also belong to the class of medications referred to as local anesthetics. In this review, we will consider lidocaine as the prototypical antiarrhythmic drug because it continues to be widely used both as an antiarrhythmic drug (first used as an antiarrhythmic drug in 1950) as well as a local anesthetic agent. Both of these clinical uses depend upon block of sodium current (I(Na)), but it is the presence of use-dependent I(Na) block, that is, an increasing amount of block at faster heart rates, which enables a local anesthetic agent to be a useful antiarrhythmic drug. Although many early studies investigated the action of antiarrhythmic drugs on Na currents, the availability of site-directed mutant Na channels has enabled for major advances in understanding their mechanisms of action based upon molecular conformations of the Na channel.
(c) 2010 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Using lidocaine and benzocaine to link sodium channel molecular conformations to state-dependent antiarrhythmic drug affinity.Circ Res. 2009 Aug 28;105(5):492-9. doi: 10.1161/CIRCRESAHA.109.198572. Epub 2009 Aug 6. Circ Res. 2009. PMID: 19661462 Free PMC article.
-
Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels.Proc Natl Acad Sci U S A. 1996 Aug 20;93(17):9270-5. doi: 10.1073/pnas.93.17.9270. Proc Natl Acad Sci U S A. 1996. PMID: 8799190 Free PMC article.
-
Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action.Br J Pharmacol. 2006 May;148(1):16-24. doi: 10.1038/sj.bjp.0706709. Br J Pharmacol. 2006. PMID: 16520744 Free PMC article.
-
Cardiac Na+ channels as therapeutic targets for antiarrhythmic agents.Handb Exp Pharmacol. 2006;(171):99-121. doi: 10.1007/3-540-29715-4_4. Handb Exp Pharmacol. 2006. PMID: 16610342 Review.
-
Probing gating mechanisms of sodium channels using pore blockers.Handb Exp Pharmacol. 2014;221:183-201. doi: 10.1007/978-3-642-41588-3_9. Handb Exp Pharmacol. 2014. PMID: 24737237 Review.
Cited by
-
Eugenol and lidocaine inhibit voltage-gated Na+ channels from dorsal root ganglion neurons with different mechanisms.Front Pharmacol. 2024 Jun 26;15:1354737. doi: 10.3389/fphar.2024.1354737. eCollection 2024. Front Pharmacol. 2024. PMID: 38989141 Free PMC article.
-
Lidocaine induces apoptosis in head and neck squamous cell carcinoma through activation of bitter taste receptor T2R14.Cell Rep. 2023 Dec 26;42(12):113437. doi: 10.1016/j.celrep.2023.113437. Epub 2023 Nov 22. Cell Rep. 2023. PMID: 37995679 Free PMC article.
-
Important Role of Asparagines in Coupling the Pore and Votage-Sensor Domain in Voltage-Gated Sodium Channels.Biophys J. 2015 Dec 1;109(11):2277-86. doi: 10.1016/j.bpj.2015.10.012. Biophys J. 2015. PMID: 26636939 Free PMC article.
-
Differential Inhibition by Cenobamate of Canonical Human Nav1.5 Ion Channels and Several Point Mutants.Int J Mol Sci. 2025 Jan 3;26(1):358. doi: 10.3390/ijms26010358. Int J Mol Sci. 2025. PMID: 39796214 Free PMC article.
-
Inhibitory Effects of Cenobamate on Multiple Human Cardiac Ion Channels and Possible Arrhythmogenic Consequences.Biomolecules. 2024 Dec 11;14(12):1582. doi: 10.3390/biom14121582. Biomolecules. 2024. PMID: 39766288 Free PMC article.
References
-
- Ahern CA, Eastwood AL, Dougherty DA, Horn R. Electrostatic contributions of aromatic residues in the local anesthetic receptor of voltage-gated sodium channels. Circ Res. 2008;102:86–94. - PubMed
-
- Butterworth JF, Strichartz GR. Molecular mechanisms of local anesthesia - a Review. Anesthesiology. 1990;72:711–734. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
