

Folding and finding RNA secondary structure
- PMID: 20685845
- PMCID: PMC2982177
- DOI: 10.1101/cshperspect.a003665
Folding and finding RNA secondary structure
Abstract
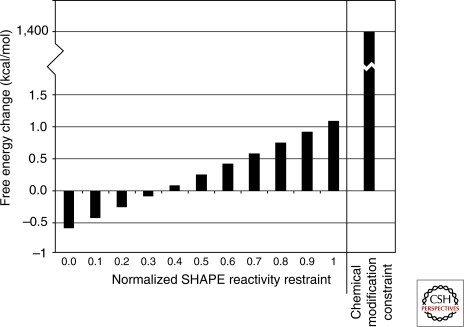
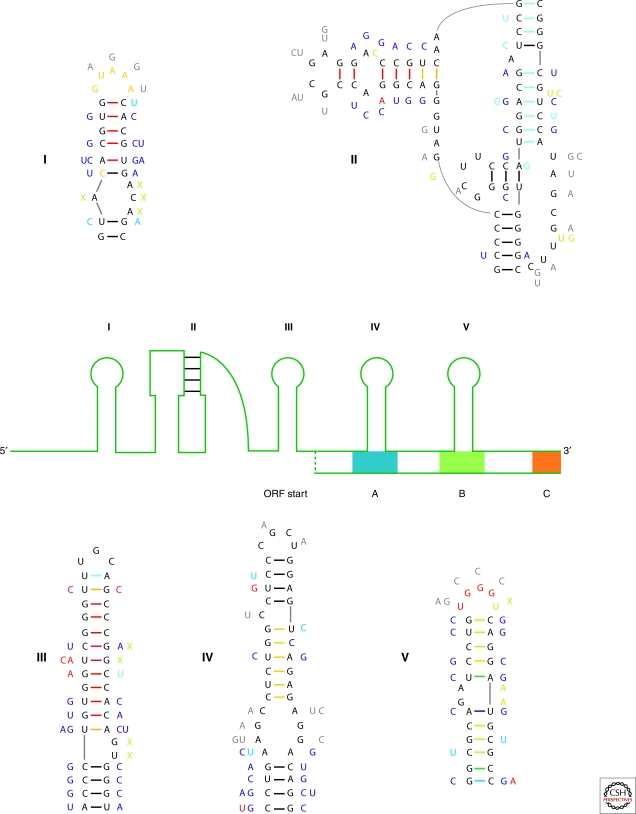
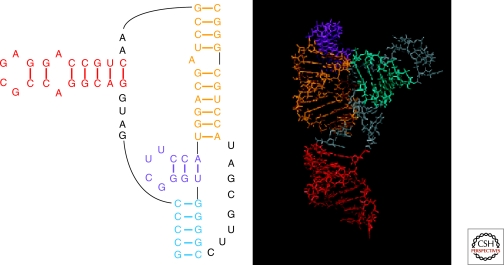
Optimal exploitation of the expanding database of sequences requires rapid finding and folding of RNAs. Methods are reviewed that automate folding and discovery of RNAs with algorithms that couple thermodynamics with chemical mapping, NMR, and/or sequence comparison. New functional noncoding RNAs in genome sequences can be found by combining sequence comparison with the assumption that functional noncoding RNAs will have more favorable folding free energies than other RNAs. When a new RNA is discovered, experiments and sequence comparison can restrict folding space so that secondary structure can be rapidly determined with the help of predicted free energies. In turn, secondary structure restricts folding in three dimensions, which allows modeling of three-dimensional structure. An example from a domain of a retrotransposon is described. Discovery of new RNAs and their structures will provide insights into evolution, biology, and design of therapeutics. Applications to studies of evolution are also reviewed.
Figures
Similar articles
-
Sequence and structure analysis of noncoding RNAs.Methods Mol Biol. 2010;609:285-306. doi: 10.1007/978-1-60327-241-4_17. Methods Mol Biol. 2010. PMID: 20221926 Review.
-
A method for aligning RNA secondary structures and its application to RNA motif detection.BMC Bioinformatics. 2005 Apr 7;6:89. doi: 10.1186/1471-2105-6-89. BMC Bioinformatics. 2005. PMID: 15817128 Free PMC article.
-
Computational analysis of noncoding RNAs.Wiley Interdiscip Rev RNA. 2012 Nov-Dec;3(6):759-78. doi: 10.1002/wrna.1134. Epub 2012 Sep 18. Wiley Interdiscip Rev RNA. 2012. PMID: 22991327 Free PMC article. Review.
-
Energy-based RNA consensus secondary structure prediction in multiple sequence alignments.Methods Mol Biol. 2014;1097:125-41. doi: 10.1007/978-1-62703-709-9_7. Methods Mol Biol. 2014. PMID: 24639158 Review.
-
Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome.Nat Biotechnol. 2005 Nov;23(11):1383-90. doi: 10.1038/nbt1144. Nat Biotechnol. 2005. PMID: 16273071
Cited by
-
DecoyFinder: Identification of Contaminants in Sets of Homologous RNA Sequences.bioRxiv [Preprint]. 2024 Oct 15:2024.10.12.618037. doi: 10.1101/2024.10.12.618037. bioRxiv. 2024. PMID: 39464058 Free PMC article. Preprint.
-
Testing the nearest neighbor model for canonical RNA base pairs: revision of GU parameters.Biochemistry. 2012 Apr 24;51(16):3508-22. doi: 10.1021/bi3002709. Epub 2012 Apr 10. Biochemistry. 2012. PMID: 22490167 Free PMC article.
-
The Potential Role of RNA Structure in Crop Molecular Breeding.Front Plant Sci. 2022 May 2;13:868771. doi: 10.3389/fpls.2022.868771. eCollection 2022. Front Plant Sci. 2022. PMID: 35586218 Free PMC article. Review.
-
The RNA encoding the microtubule-associated protein tau has extensive structure that affects its biology.PLoS One. 2019 Jul 10;14(7):e0219210. doi: 10.1371/journal.pone.0219210. eCollection 2019. PLoS One. 2019. PMID: 31291322 Free PMC article.
-
Comparative and integrative analysis of RNA structural profiling data: current practices and emerging questions.Quant Biol. 2017 Mar;5(1):3-24. doi: 10.1007/s40484-017-0093-6. Epub 2017 Mar 30. Quant Biol. 2017. PMID: 28717530 Free PMC article.
References
-
- Alvarez I, Wendel JF 2003. Ribosomal ITS sequences and plant phylogenetic inference. Mol Phylogenet Evol 29: 417–434 - PubMed
-
- Andronescu M, Condon A, Hoos HH, Mathews DH, Murphy KP 2007. Efficient parameter estimation for RNA secondary structure prediction. Bioinformatics 23: i19–i28 - PubMed
-
- Bartel DP 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297 - PubMed
-
- Berezikov E, Cuppen E, Plasterk RH 2006. Approaches to microRNA discovery. Nat Genet 38Suppl: S2–7 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources