

Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into huntington's disease molecular pathogenesis
- PMID: 20685997
- PMCID: PMC3078518
- DOI: 10.1523/JNEUROSCI.0146-10.2010
Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into huntington's disease molecular pathogenesis
Abstract
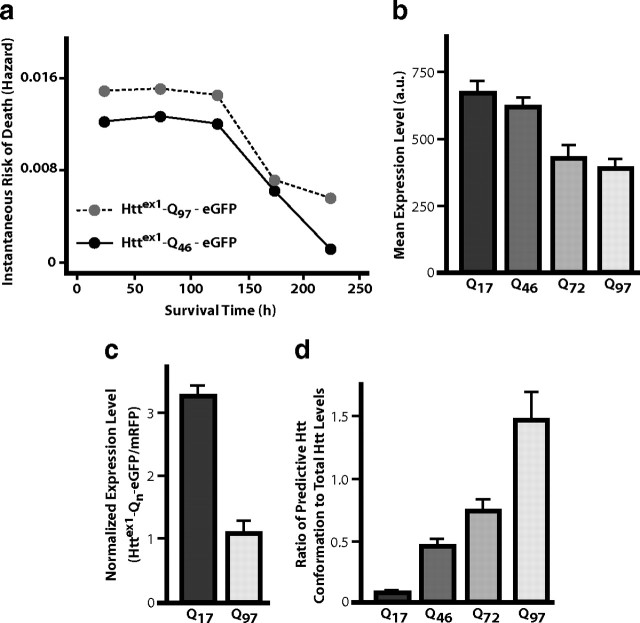
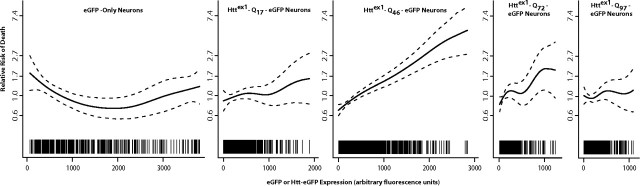
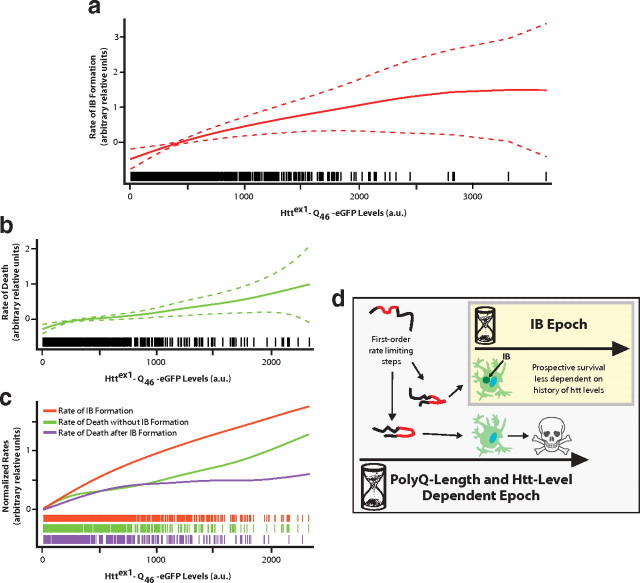
An expanded polyglutamine (polyQ) stretch in the protein huntingtin (htt) induces self-aggregation into inclusion bodies (IBs) and causes Huntington's disease (HD). Defining precise relationships between early observable variables and neuronal death at the molecular and cellular levels should improve our understanding of HD pathogenesis. Here, we used an automated microscope that tracks thousands of neurons individually over their entire lifetime to quantify interconnected relationships between early variables, such as htt levels, polyQ length, and IB formation, and neuronal death in a primary striatal model of HD. The resulting model revealed that mutant htt increases the risk of death by tonically interfering with homeostatic coping mechanisms rather than producing accumulated damage to the neuron, htt toxicity is saturable, the rate-limiting steps for inclusion body formation and death can be traced to different conformational changes in monomeric htt, and IB formation reduces the impact of the starting levels of htt of a neuron on its risk of death. Finally, the model that emerges from our quantitative measurements places critical limits on the potential mechanisms by which mutant htt might induce neurodegeneration, which should help direct future research.
Figures

References
-
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. - PubMed
-
- Benn CL, Landles C, Li H, Strand AD, Woodman B, Sathasivam K, Li SH, Ghazi-Noori S, Hockly E, Faruque SM, Cha JH, Sharpe PT, Olson JM, Li XJ, Bates GP. Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington's disease. Hum Mol Genet. 2005;14:3065–3078. - PubMed
-
- Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17:351–365. - PubMed
-
- Brooks E, Arrasate M, Cheung K, Finkbeiner SM. Using antibodies to analyze polyglutamine stretches. Methods Mol Biol. 2004;277:103–128. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- AG11385/AG/NIA NIH HHS/United States
- R01 AG011385/AG/NIA NIH HHS/United States
- 2R01 NS045091/NS/NINDS NIH HHS/United States
- R01 NS039074/NS/NINDS NIH HHS/United States
- 2R01 NS039074/NS/NINDS NIH HHS/United States
- 2P01 AG022074/AG/NIA NIH HHS/United States
- R01 AG018440/AG/NIA NIH HHS/United States
- R01 NS045191/NS/NINDS NIH HHS/United States
- R37 AG018440/AG/NIA NIH HHS/United States
- AG022074/AG/NIA NIH HHS/United States
- R01 NS045091/NS/NINDS NIH HHS/United States
- AG18440/AG/NIA NIH HHS/United States
- P01 AG022074/AG/NIA NIH HHS/United States
- R37 AG011385/AG/NIA NIH HHS/United States
- C06 RR018928/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical