

Long-term restriction by APOBEC3F selects human immunodeficiency virus type 1 variants with restored Vif function
- PMID: 20686027
- PMCID: PMC2937771
- DOI: 10.1128/JVI.00632-10
Long-term restriction by APOBEC3F selects human immunodeficiency virus type 1 variants with restored Vif function
Abstract
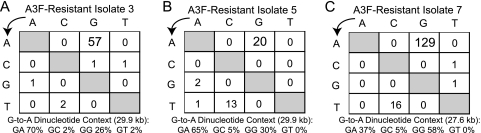
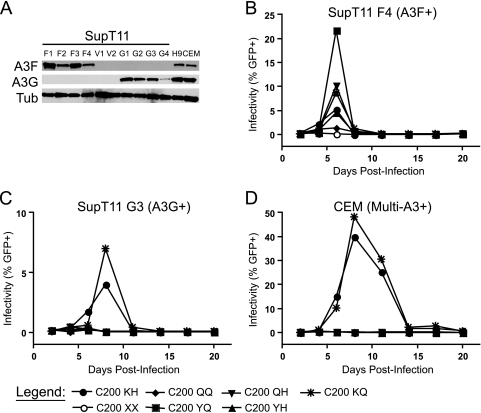
Tandem stop mutations K26X and H27X in human immunodeficiency virus type 1 (HIV-1) vif compromise virus replication in human T-cell lines that stably express APOBEC3F (A3F) or APOBEC3G (A3G). We previously reported that partial resistance to A3G could develop in these Vif-deficient viruses through a nucleotide A200-to-T/C transversion and a vpr null mutation, but these isolates were still susceptible to restriction by A3F. Here, long-term selection experiments were done to determine how these A3G-selected isolates might evolve to spread in the presence of A3F. We found that A3F, like A3G, is capable of potent, long-term restriction that eventually selects for heritable resistance. In all 7 instances, the selected isolates had restored Vif function to cope with A3F activity. In two isolates, Vif Q26-Q27 and Y26-Q27, the resistance phenotype recapitulated in molecular clones, but when the selected vif alleles were analyzed in the context of an otherwise wild-type viral background, a different outcome emerged. Although HIV-1 clones with Vif Q26-Q27 or Y26-Q27 were fully capable of overcoming A3F, they were now susceptible to restriction by A3G. Concordant with prior studies, a lysine at position 26 proved essential for A3G neutralization. These data combine to indicate that A3F and A3G exert at least partly distinct selective pressures and that Vif function may be essential for the virus to replicate in the presence of A3F.
Figures





References
-
- Abudu, A., A. Takaori-Kondo, T. Izumi, K. Shirakawa, M. Kobayashi, A. Sasada, K. Fukunaga, and T. Uchiyama. 2006. Murine retrovirus escapes from murine APOBEC3 via two distinct novel mechanisms. Curr. Biol. 16:1565-1570. - PubMed
-
- Bishop, K. N., R. K. Holmes, A. M. Sheehy, N. O. Davidson, S. J. Cho, and M. H. Malim. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14:1392-1396. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
