

Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling
- PMID: 20686047
- PMCID: PMC2950581
- DOI: 10.1128/JVI.00949-10
Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling
Abstract
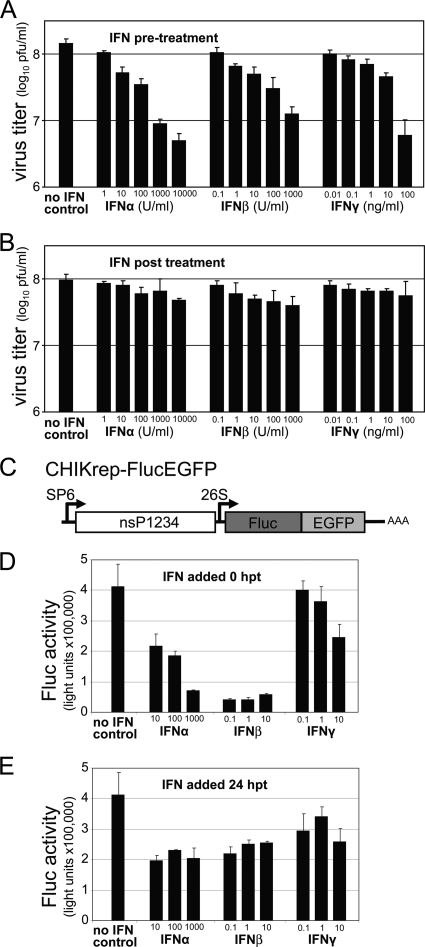
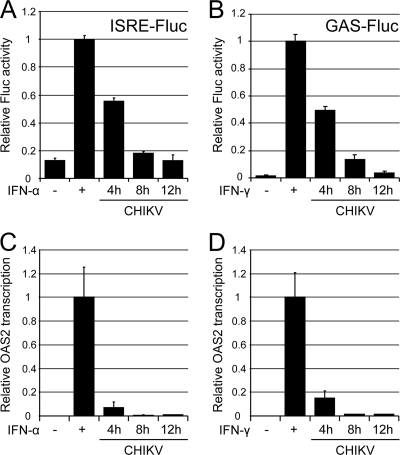
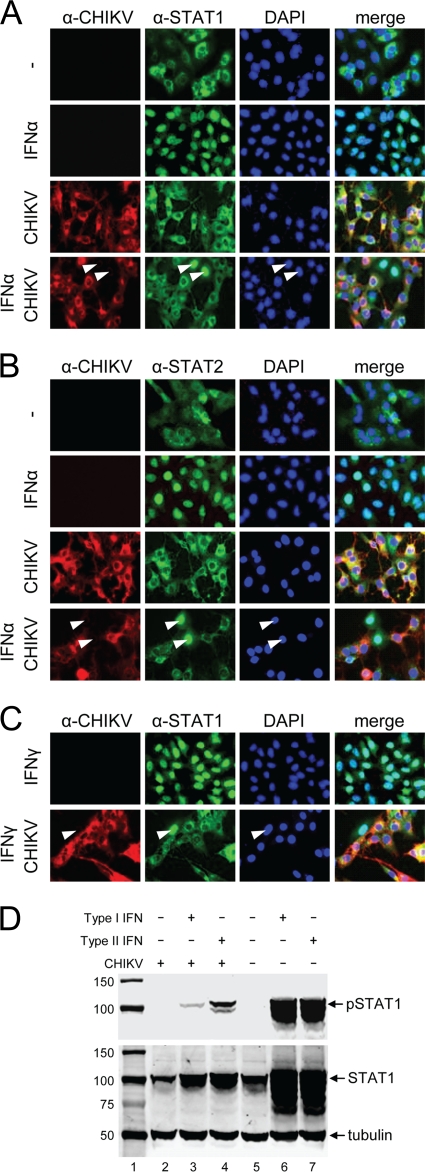
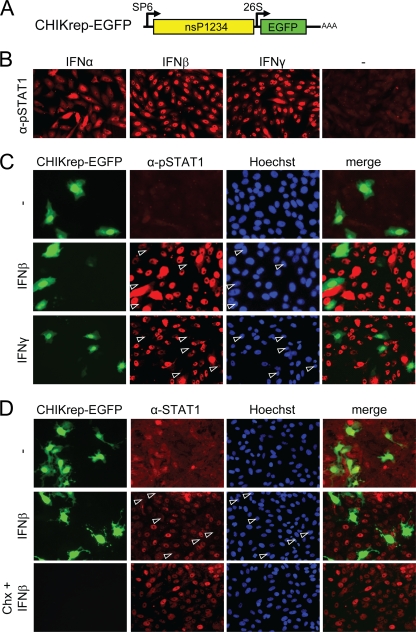
Chikungunya virus (CHIKV) is an emerging human pathogen transmitted by mosquitoes. Like that of other alphaviruses, CHIKV replication causes general host shutoff, leading to severe cytopathicity in mammalian cells, and inhibits the ability of infected cells to respond to interferon (IFN). Recent research, however, suggests that alphaviruses may have additional mechanisms to circumvent the host's antiviral IFN response. Here we show that CHIKV replication is resistant to inhibition by interferon once RNA replication has been established and that CHIKV actively suppresses the antiviral IFN response by preventing IFN-induced gene expression. Both CHIKV infection and CHIKV replicon RNA replication efficiently blocked STAT1 phosphorylation and/or nuclear translocation in mammalian cells induced by either type I or type II IFN. Expression of individual CHIKV nonstructural proteins (nsPs) showed that nsP2 was a potent inhibitor of IFN-induced JAK-STAT signaling. In addition, mutations in CHIKV-nsP2 (P718S) and Sindbis virus (SINV)-nsP2 (P726S) that render alphavirus replicons noncytopathic significantly reduced JAK-STAT inhibition. This host shutoff-independent inhibition of IFN signaling by CHIKV is likely to have an important role in viral pathogenesis.
Figures
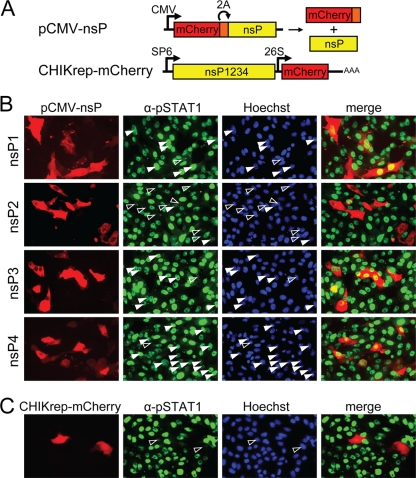
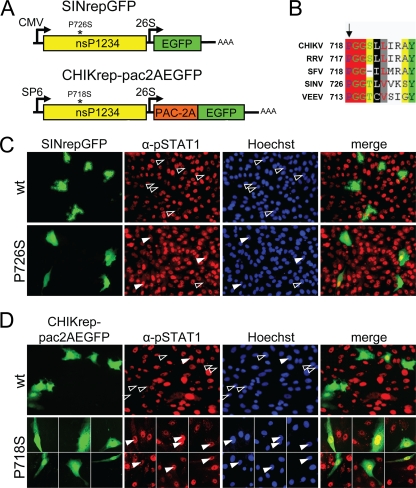
References
-
- Couderc, T., F. Chretien, C. Schilte, O. Disson, M. Brigitte, F. Guivel-Benhassine, Y. Touret, G. Barau, N. Cayet, I. Schuffenecker, P. Despres, F. Arenzana-Seisdedos, A. Michault, M. L. Albert, and M. Lecuit. 2008. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 4:e29. - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
