

Genomic signatures of germline gene expression
- PMID: 20686123
- PMCID: PMC2963814
- DOI: 10.1101/gr.106666.110
Genomic signatures of germline gene expression
Abstract
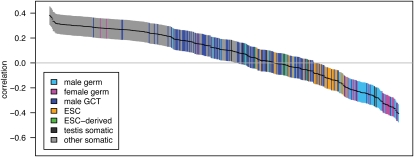
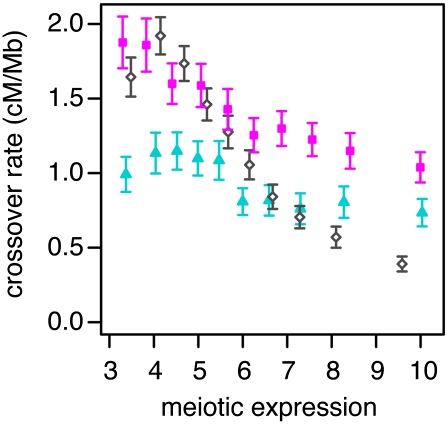
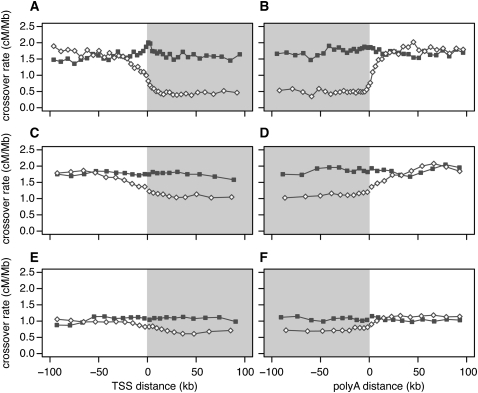
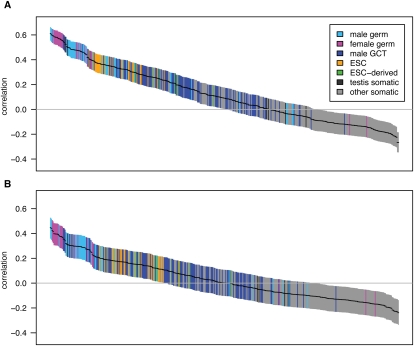
Transcribed regions in the human genome differ from adjacent intergenic regions in transposable element density, crossover rates, and asymmetric substitution and sequence composition patterns. We tested whether these differences reflect selection or are instead a byproduct of germline transcription, using publicly available gene expression data from a variety of germline and somatic tissues. Crossover rate shows a strong negative correlation with gene expression in meiotic tissues, suggesting that crossover is inhibited by transcription. Strand-biased composition (G+T content) and A → G versus T → C substitution asymmetry are both positively correlated with germline gene expression. We find no evidence for a strand bias in allele frequency data, implying that the substitution asymmetry reflects a mutation rather than a fixation bias. The density of transposable elements is positively correlated with germline expression, suggesting that such elements preferentially insert into regions that are actively transcribed. For each of the features examined, our analyses favor a nonselective explanation for the observed trends and point to the role of germline gene expression in shaping the mammalian genome.
Figures
References
-
- Bolstad BM, Irizarry RA, Astrand M, Speed TP 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources