

Predicting protein structures with a multiplayer online game
- PMID: 20686574
- PMCID: PMC2956414
- DOI: 10.1038/nature09304
Predicting protein structures with a multiplayer online game
Abstract
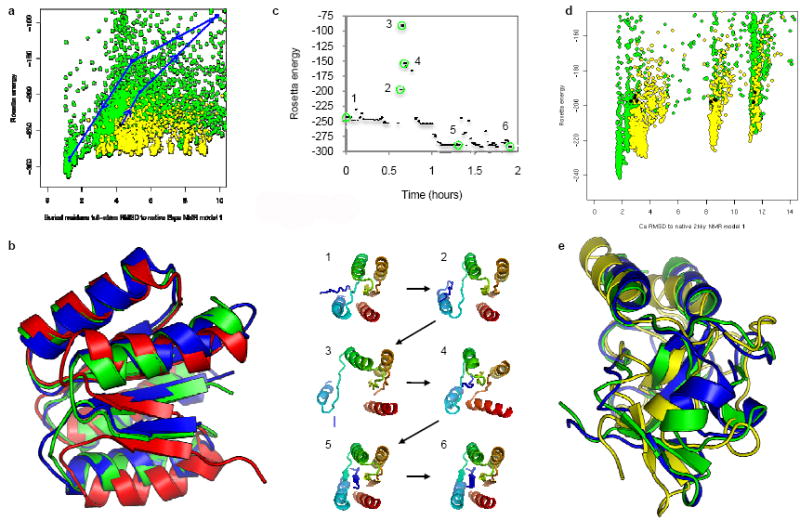
People exert large amounts of problem-solving effort playing computer games. Simple image- and text-recognition tasks have been successfully 'crowd-sourced' through games, but it is not clear if more complex scientific problems can be solved with human-directed computing. Protein structure prediction is one such problem: locating the biologically relevant native conformation of a protein is a formidable computational challenge given the very large size of the search space. Here we describe Foldit, a multiplayer online game that engages non-scientists in solving hard prediction problems. Foldit players interact with protein structures using direct manipulation tools and user-friendly versions of algorithms from the Rosetta structure prediction methodology, while they compete and collaborate to optimize the computed energy. We show that top-ranked Foldit players excel at solving challenging structure refinement problems in which substantial backbone rearrangements are necessary to achieve the burial of hydrophobic residues. Players working collaboratively develop a rich assortment of new strategies and algorithms; unlike computational approaches, they explore not only the conformational space but also the space of possible search strategies. The integration of human visual problem-solving and strategy development capabilities with traditional computational algorithms through interactive multiplayer games is a powerful new approach to solving computationally-limited scientific problems.
Figures



References
-
- von Ahn L, Dabbish L. CHI ’04: Proceedings of the 2004 Conference on Human Factors in Computing Systems. ACM Press; 2004. Labeling images with a computer game; pp. 319–326. URL http://dx.doi.org/10.1145/985692.985733. - DOI
-
- von Ahn L, Liu R, Blum M. CHI ’06: Proceedings of the SIGCHI Conference on Human Factors in Computing Systems. ACM; New York, NY, USA: 2006. Peekaboom: a game for locating objects in images; pp. 55–64. URL http://dx.doi.org/10.1145/1124772.1124782. - DOI
-
- Westphal AJ, et al. Non-destructive search for interstellar dust using synchrotron microprobes. Proceeding of ICXOM20, 20th International Congress on X-ray Optics Microanalysis
-
- Rohl C, Strauss C, Misura K, Baker D. Methods in Enzymology. Vol. 383. Department of Biochemistry and Howard Hughes Medical Institute, University of Washington, Seattle; Washington 98195, USA: 2004. Protein structure prediction using Rosetta; pp. 66–93. URL http://dx.doi.org/10.1016/S0076-6879(04)83004-0. - DOI - PubMed
-
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. URL http://view.ncbi.nlm.nih.gov/pubmed/73220860. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
