

Nuclear factor-kappa B family member RelB inhibits human immunodeficiency virus-1 Tat-induced tumor necrosis factor-alpha production
- PMID: 20686703
- PMCID: PMC2912378
- DOI: 10.1371/journal.pone.0011875
Nuclear factor-kappa B family member RelB inhibits human immunodeficiency virus-1 Tat-induced tumor necrosis factor-alpha production
Abstract
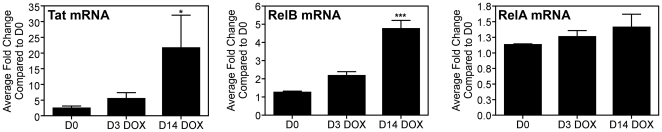
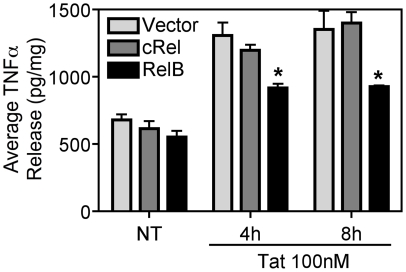
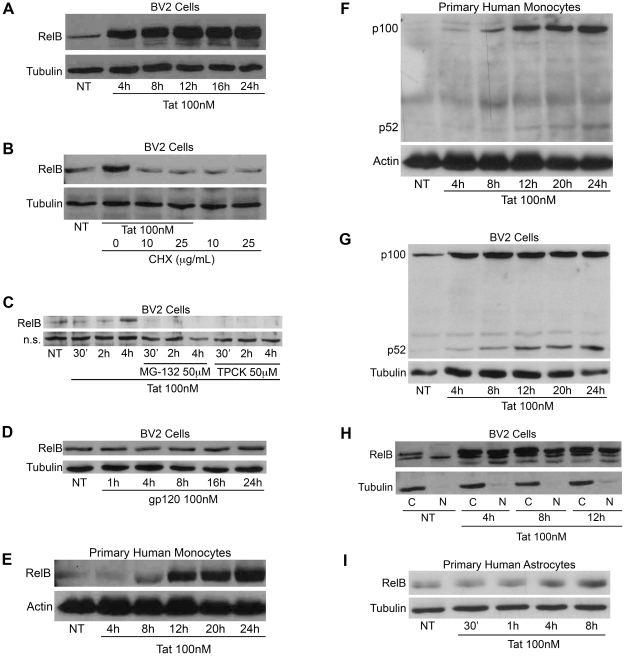
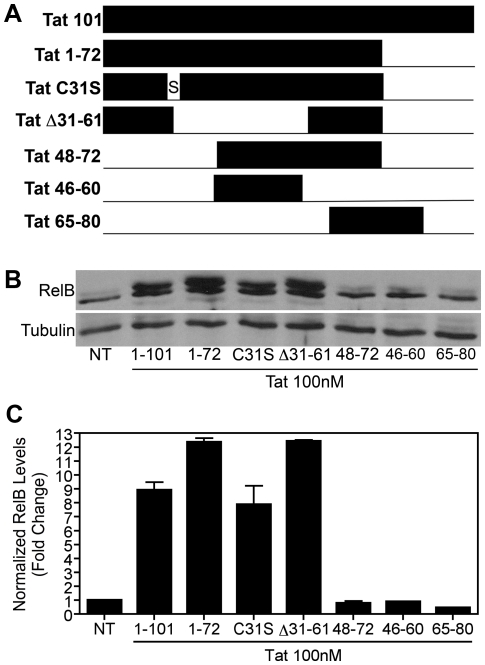
Human Immunodeficiency Virus-1 (HIV-1)-associated neurocognitive disorder (HAND) is likely neuroinflammatory in origin, believed to be triggered by inflammatory and oxidative stress responses to cytokines and HIV protein gene products such as the HIV transactivator of transcription (Tat). Here we demonstrate increased messenger RNA for nuclear factor-kappa B (NF-kappaB) family member, transcription factor RelB, in the brain of doxycycline-induced Tat transgenic mice, and increased RelB synthesis in Tat-exposed microglial cells. Since genetic ablation of RelB in mice leads to multi-organ inflammation, we hypothesized that Tat-induced, newly synthesized RelB inhibits cytokine production by microglial cells, possibly through the formation of transcriptionally inactive RelB/RelA complexes. Indeed, tumor necrosis factor-alpha (TNFalpha) production in monocytes isolated from RelB deficient mice was significantly higher than in monocytes isolated from RelB expressing controls. Moreover, RelB overexpression in microglial cells inhibited Tat-induced TNFalpha synthesis in a manner that involved transcriptional repression of the TNFalpha promoter, and increased phosphorylation of RelA at serine 276, a prerequisite for increased RelB/RelA protein interactions. The Rel-homology-domain within RelB was necessary for this interaction. Overexpression of RelA itself, in turn, significantly increased TNFalpha promoter activity, an effect that was completely blocked by RelB overexpression. We conclude that RelB regulates TNFalpha cytokine synthesis by competitive interference binding with RelA, which leads to downregulation of TNFalpha production. Moreover, because Tat activates both RelB and TNFalpha in microglia, and because Tat induces inflammatory TNFalpha synthesis via NF-kappaB, we posit that RelB serves as a cryoprotective, anti-inflammatory, counter-regulatory mechanism for pathogenic NF-kappaB activation. These findings identify a novel regulatory pathway for controlling HIV-induced microglial activation and cytokine production that may have important therapeutic implications for the management of HAND.
Conflict of interest statement
Figures




References
-
- Resnick L, Berger JR, Shapshak P, Tourtellotte WW. Early penetration of the blood-brain-barrier by HIV. Neurology. 1988;38:9–14. - PubMed
-
- Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4:307–318. - PubMed
-
- Masliah E, DeTeresa RM, Mallory ME, Hansen LA. Changes in pathological findings at autopsy in AIDS cases for the last 15 years. AIDS. 2000;14:69–74. - PubMed
-
- Nath A, Sacktor N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr Opin Neurol. 2006;19:358–361. - PubMed
-
- Tozzi V, Balestra P, Bellagamba R, Corpolongo A, Salvatori MF, et al. Persistence of neuropsychologic deficits despite long-term highly active antiretroviral therapy in patients with HIV-related neurocognitive impairment: prevalence and risk factors. J Acquir Immune Defic Syndr. 2007;45:174–182. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
