

Congruency between biophysical data from multiple platforms and molecular dynamics simulation of the double-super helix model of nascent high-density lipoprotein
- PMID: 20687589
- PMCID: PMC2940317
- DOI: 10.1021/bi100588a
Congruency between biophysical data from multiple platforms and molecular dynamics simulation of the double-super helix model of nascent high-density lipoprotein
Abstract
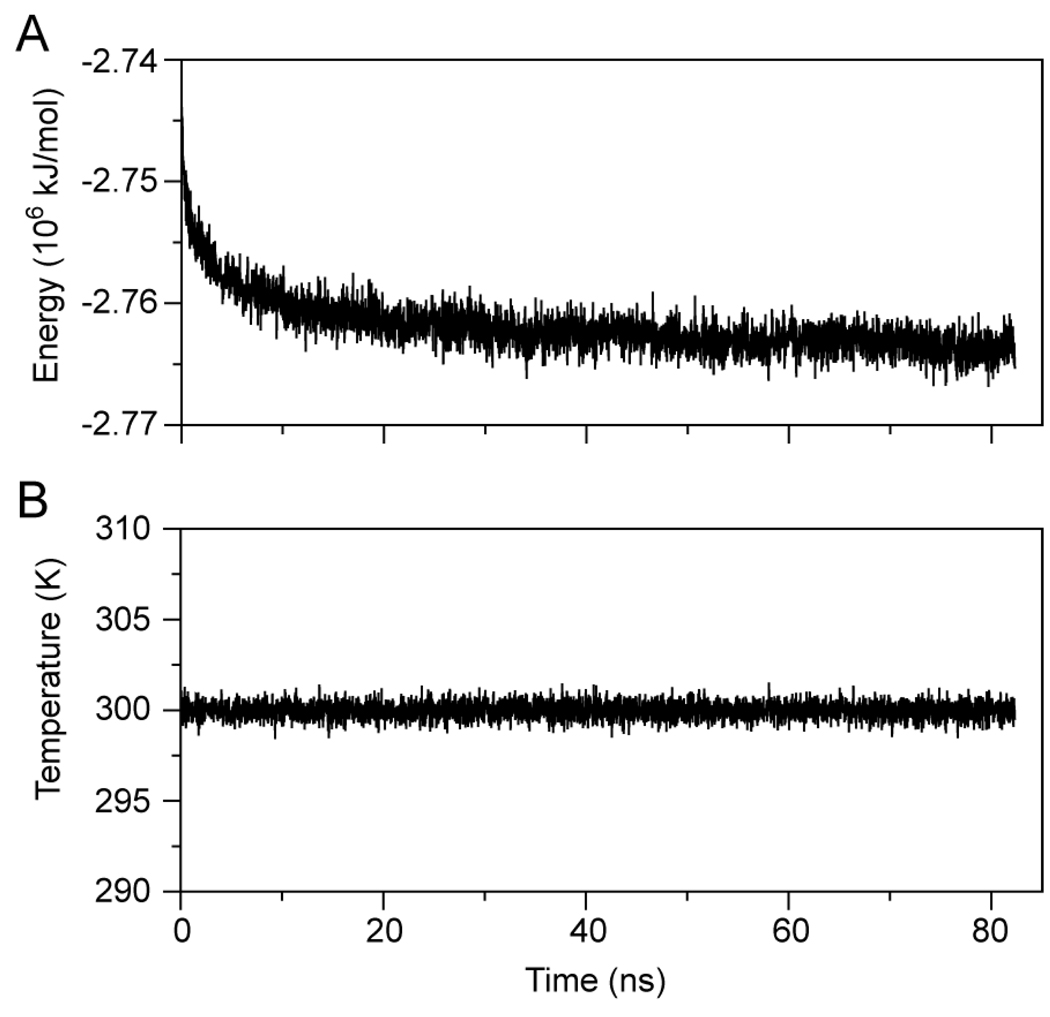
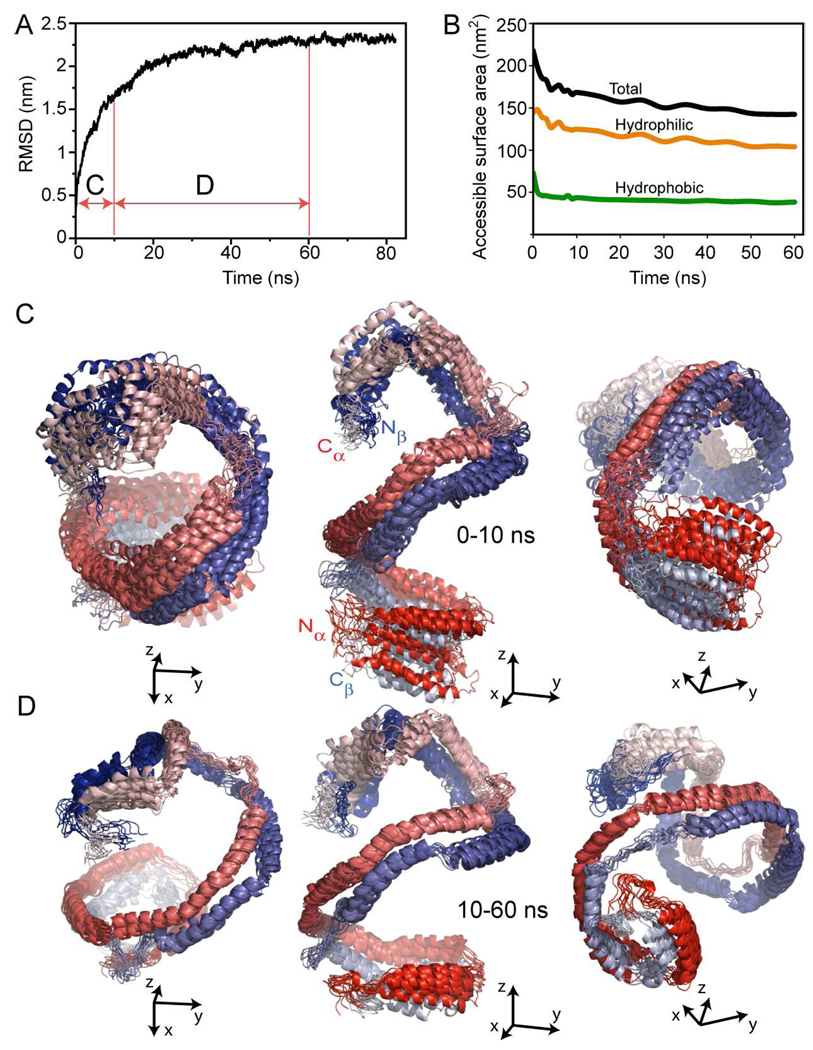
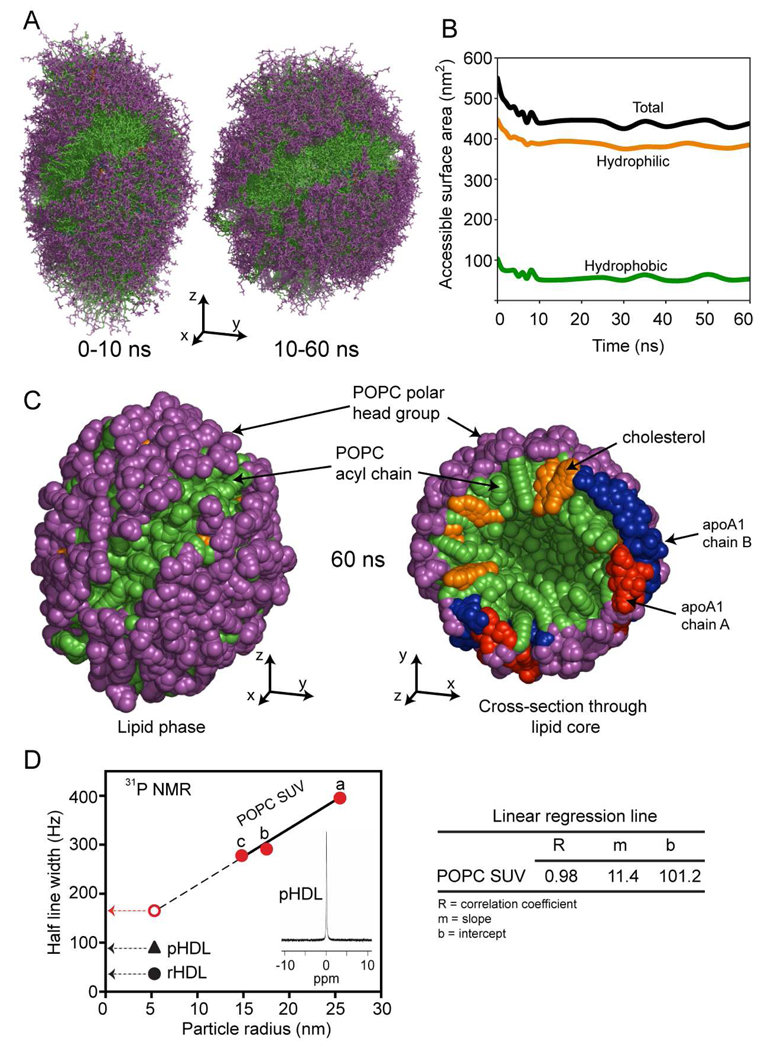
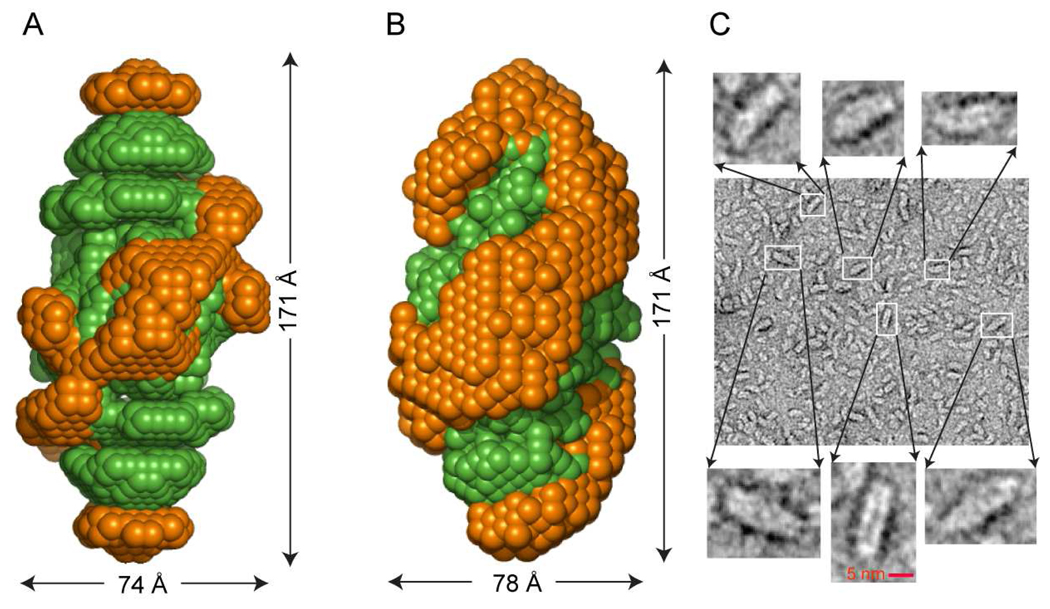
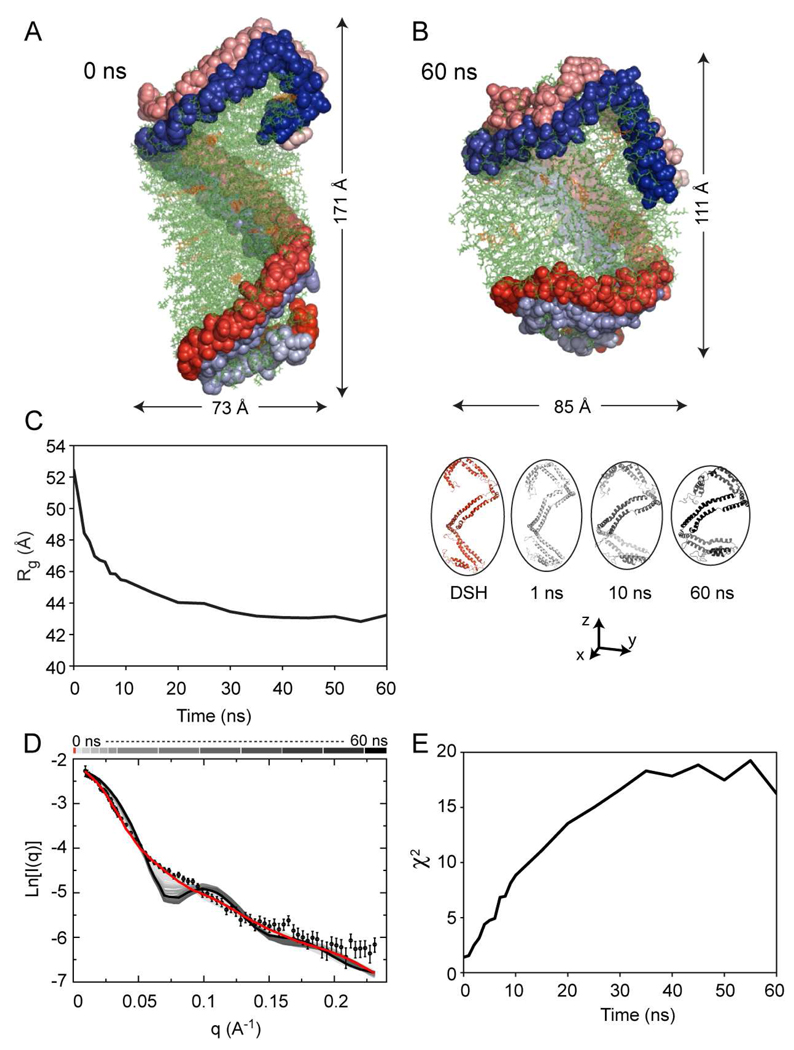
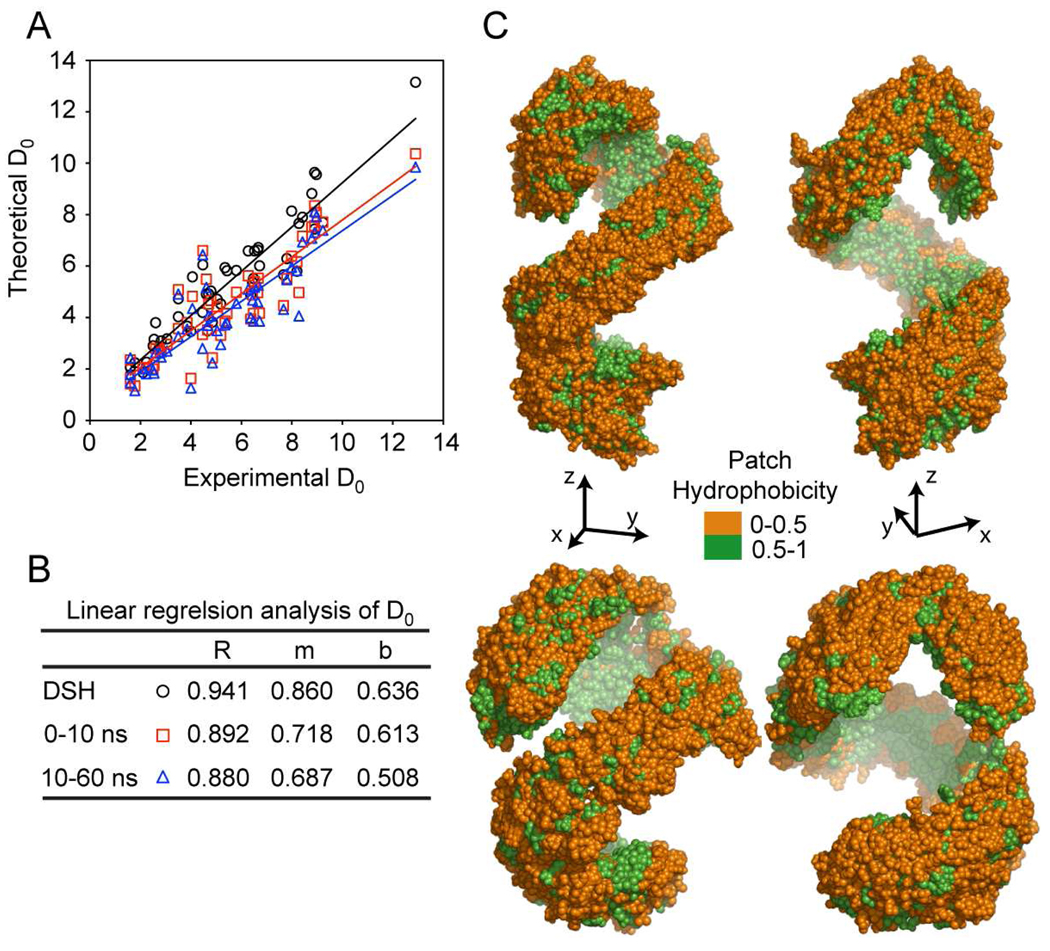
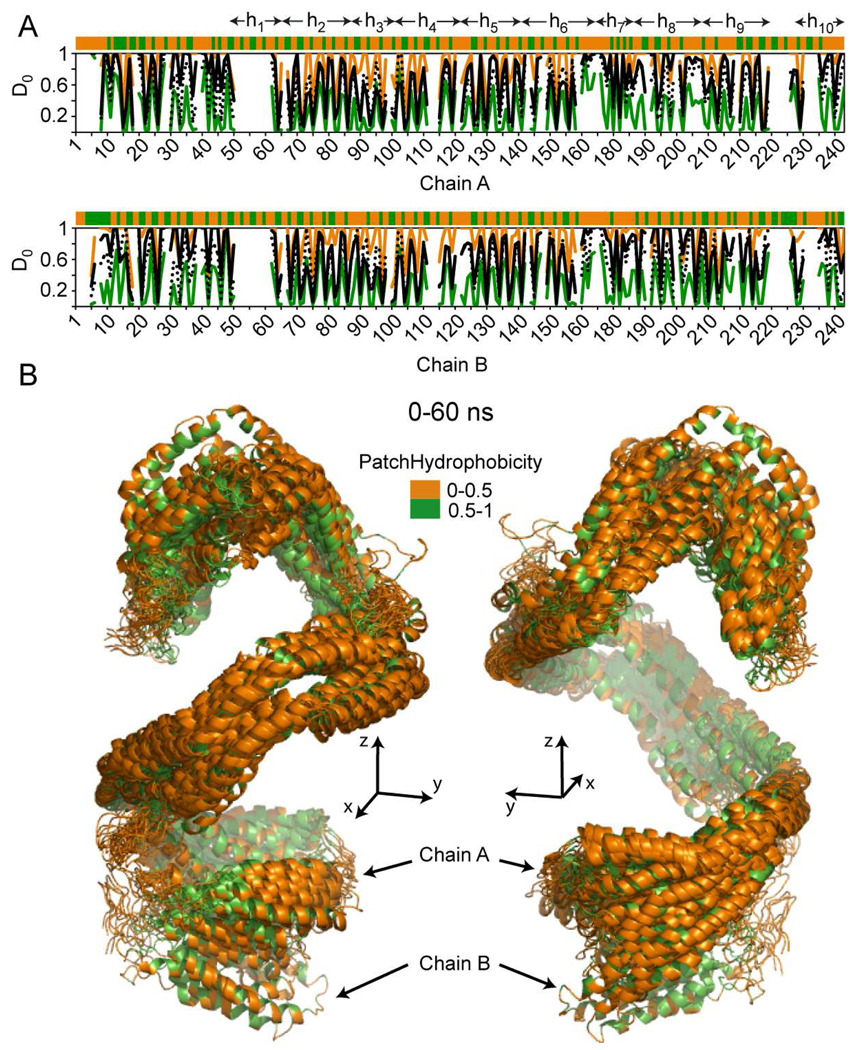
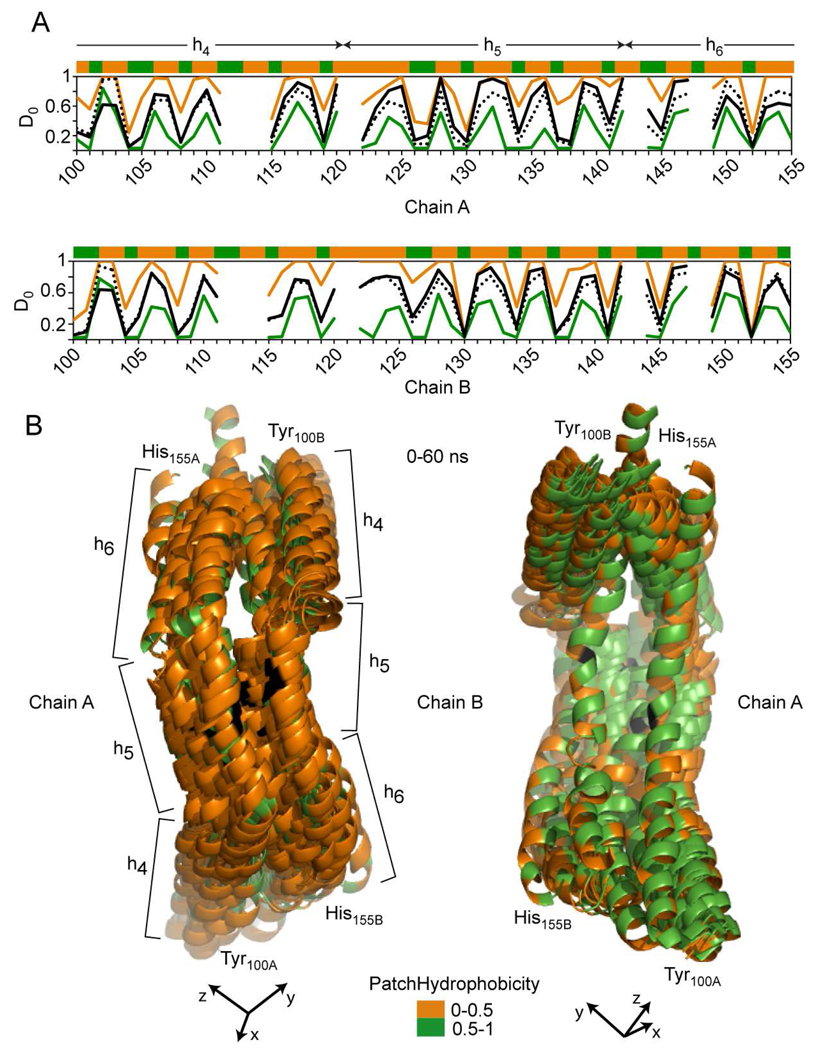
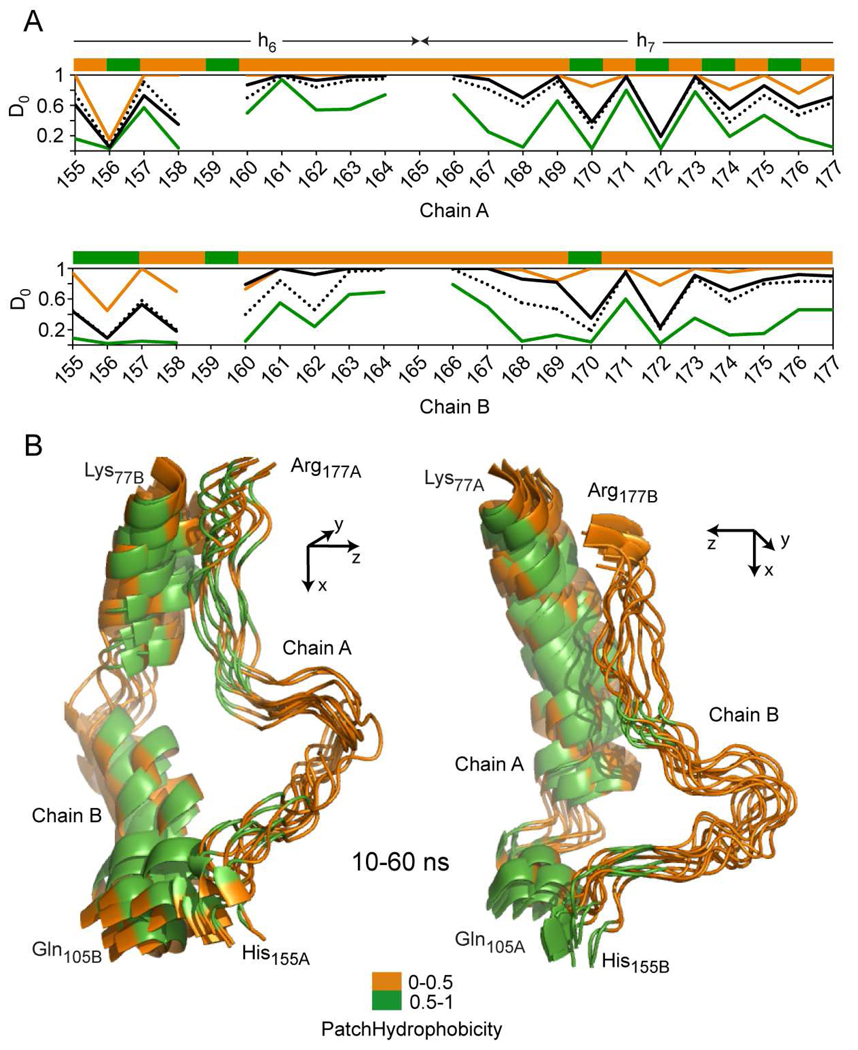
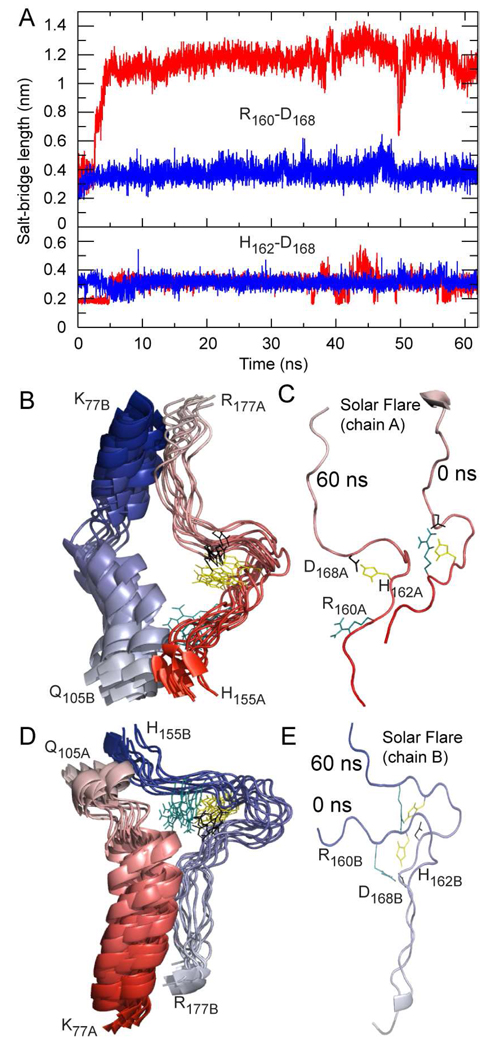
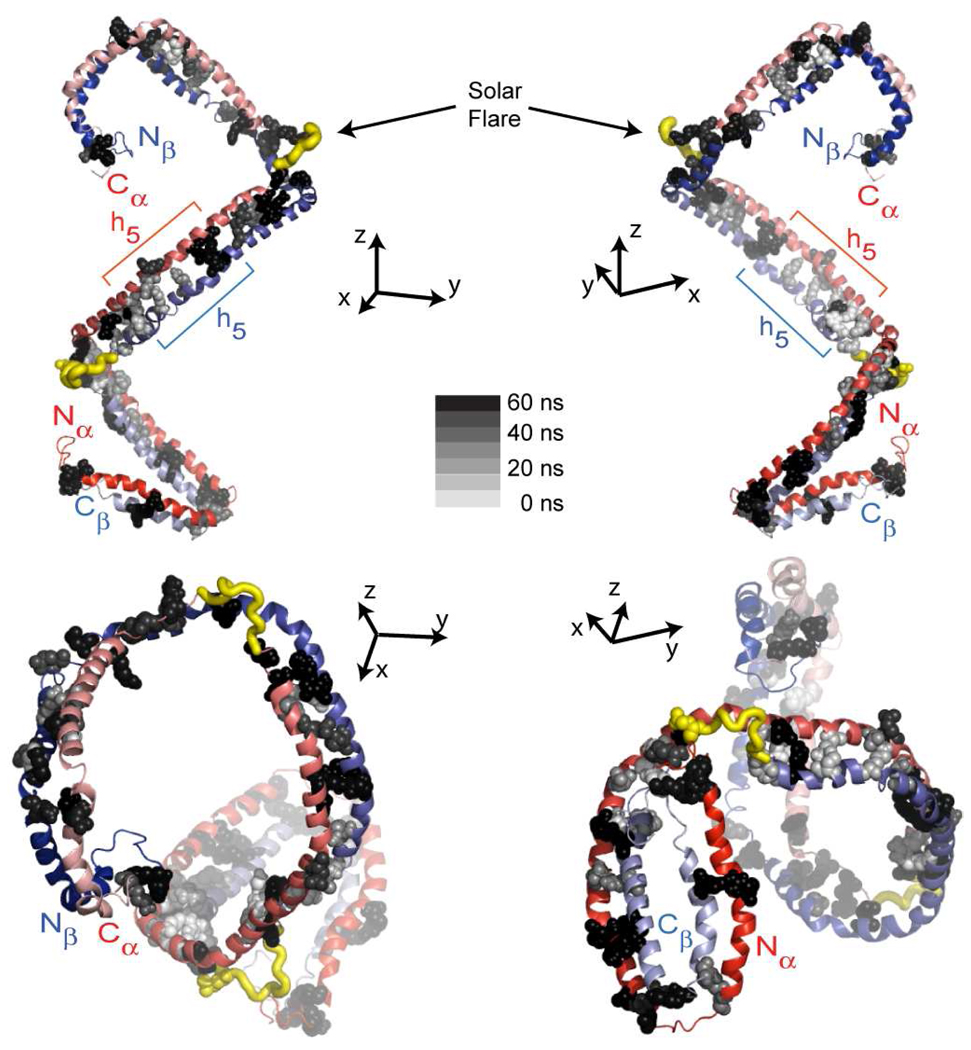
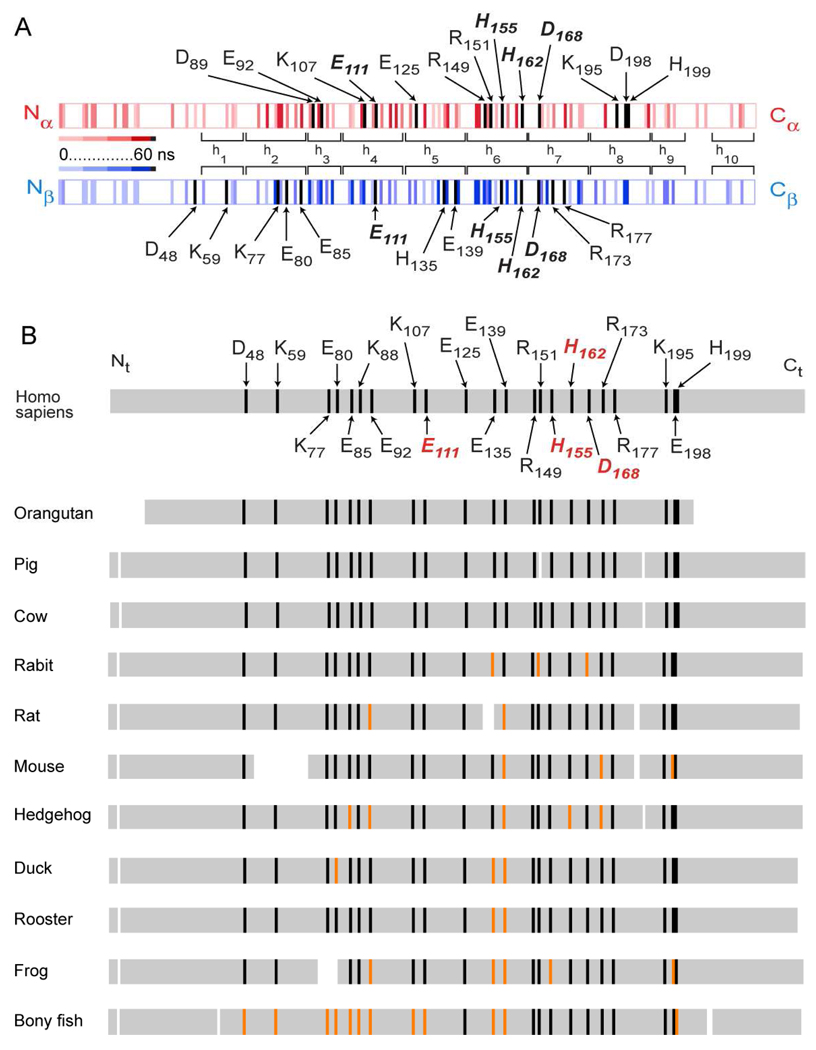
The predicted structure and molecular trajectories from >80 ns molecular dynamics simulation of the solvated Double-Super Helix (DSH) model of nascent high-density lipoprotein (HDL) were determined and compared with experimental data on reconstituted nascent HDL obtained from multiple biophysical platforms, including small angle neutron scattering (SANS) with contrast variation, hydrogen-deuterium exchange tandem mass spectrometry (H/D-MS/MS), nuclear magnetic resonance spectroscopy (NMR), cross-linking tandem mass spectrometry (MS/MS), fluorescence resonance energy transfer (FRET), electron spin resonance spectroscopy (ESR), and electron microscopy. In general, biophysical constraints experimentally derived from the multiple platforms agree with the same quantities evaluated using the simulation trajectory. Notably, key structural features postulated for the recent DSH model of nascent HDL are retained during the simulation, including (1) the superhelical conformation of the antiparallel apolipoprotein A1 (apoA1) chains, (2) the lipid micellar-pseudolamellar organization, and (3) the solvent-exposed Solar Flare loops, proposed sites of interaction with LCAT (lecithin cholesteryl acyltransferase). Analysis of salt bridge persistence during simulation provides insights into structural features of apoA1 that forms the backbone of the lipoprotein. The combination of molecular dynamics simulation and experimental data from a broad range of biophysical platforms serves as a powerful approach to studying large macromolecular assemblies such as lipoproteins. This application to nascent HDL validates the DSH model proposed earlier and suggests new structural details of nascent HDL.
Figures
Similar articles
-
Protein Backbone and Average Particle Dynamics in Reconstituted Discoidal and Spherical HDL Probed by Hydrogen Deuterium Exchange and Elastic Incoherent Neutron Scattering.Biomolecules. 2020 Jan 10;10(1):121. doi: 10.3390/biom10010121. Biomolecules. 2020. PMID: 31936876 Free PMC article.
-
Double superhelix model of high density lipoprotein.J Biol Chem. 2009 Dec 25;284(52):36605-36619. doi: 10.1074/jbc.M109.039537. Epub 2009 Oct 7. J Biol Chem. 2009. PMID: 19812036 Free PMC article.
-
A Systematic Investigation of Structure/Function Requirements for the Apolipoprotein A-I/Lecithin Cholesterol Acyltransferase Interaction Loop of High-density Lipoprotein.J Biol Chem. 2016 Mar 18;291(12):6386-95. doi: 10.1074/jbc.M115.696088. Epub 2016 Jan 21. J Biol Chem. 2016. PMID: 26797122 Free PMC article.
-
Structural Insights into High Density Lipoprotein: Old Models and New Facts.Front Pharmacol. 2016 Jan 12;6:318. doi: 10.3389/fphar.2015.00318. eCollection 2015. Front Pharmacol. 2016. PMID: 26793109 Free PMC article. Review.
-
Apolipoprotein A-I: structure-function relationships.J Lipid Res. 2000 Jun;41(6):853-72. J Lipid Res. 2000. PMID: 10828078 Review.
Cited by
-
An abundant dysfunctional apolipoprotein A1 in human atheroma.Nat Med. 2014 Feb;20(2):193-203. doi: 10.1038/nm.3459. Epub 2014 Jan 26. Nat Med. 2014. PMID: 24464187 Free PMC article.
-
The low-resolution structure of nHDL reconstituted with DMPC with and without cholesterol reveals a mechanism for particle expansion.J Lipid Res. 2013 Apr;54(4):966-83. doi: 10.1194/jlr.M032763. Epub 2013 Jan 23. J Lipid Res. 2013. PMID: 23349207 Free PMC article.
-
Molecules that mimic apolipoprotein A-I: potential agents for treating atherosclerosis.J Med Chem. 2014 Mar 27;57(6):2169-96. doi: 10.1021/jm4005847. Epub 2013 Oct 29. J Med Chem. 2014. PMID: 24168751 Free PMC article. Review.
-
Coarse-Grained Conformational Sampling of Protein Structure Improves the Fit to Experimental Hydrogen-Exchange Data.Front Mol Biosci. 2017 Mar 10;4:13. doi: 10.3389/fmolb.2017.00013. eCollection 2017. Front Mol Biosci. 2017. PMID: 28344973 Free PMC article.
-
Conformational transitions in the membrane scaffold protein of phospholipid bilayer nanodiscs.Mol Cell Proteomics. 2011 Sep;10(9):M111.010876. doi: 10.1074/mcp.M111.010876. Epub 2011 Jun 29. Mol Cell Proteomics. 2011. PMID: 21715319 Free PMC article.
References
-
- Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. Cell Metab. 2008;7:365–375. - PubMed
-
- Trigatti B, Rigotti A, Krieger M. Curr. Opin. Lipidol. 2000;11:123–131. - PubMed
-
- Assmann G, Gotto AM., Jr Circulation. 2004;109:III8–III14. - PubMed
-
- Barter PJ, Rye KA. Curr. Opin. Lipidol. 2006;17:399–403. - PubMed
-
- Rader DJ. Nat. Clin. Pract. Cardiovasc. Med. 2007;4:102–109. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
