

Genetic manipulation of AML1-ETO-induced expansion of hematopoietic precursors in a Drosophila model
- PMID: 20688956
- PMCID: PMC2996118
- DOI: 10.1182/blood-2010-03-276998
Genetic manipulation of AML1-ETO-induced expansion of hematopoietic precursors in a Drosophila model
Abstract
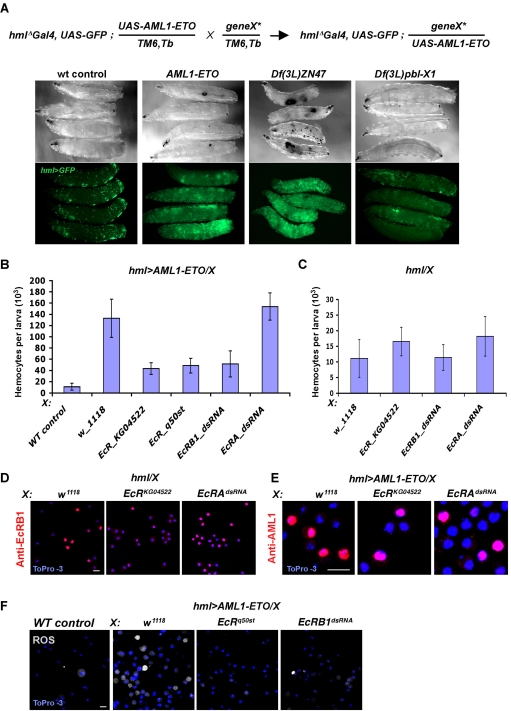
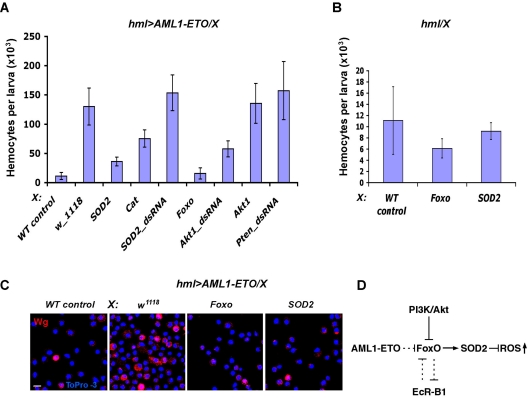
Among mutations in human Runx1/AML1 transcription factors, the t(8;21)(q22;q22) genomic translocation that creates an AML1-ETO fusion protein is implicated in etiology of the acute myeloid leukemia. To identify genes and components associated with this oncogene we used Drosophila as a genetic model. Expression of AML1-ETO caused an expansion of hematopoietic precursors in Drosophila, which expressed high levels of reactive oxygen species (ROS). Mutations in functional domains of the fusion protein suppress the proliferative phenotype. In a genetic screen, we found that inactivation of EcRB1 or activation of Foxo and superoxide dismutase-2 (SOD2) suppress the AML1-ETO-induced phenotype by reducing ROS expression in the precursor cells. Our studies indicate that ROS is a signaling factor promoting maintenance of normal as well as the aberrant myeloid precursors and suggests the importance of antioxidant enzymes and their regulators as targets for further study in the context of leukemia.
Figures


References
-
- Canon J, Banerjee U. Runt and Lozenge function in Drosophila development. Semin Cell Dev Biol. 2000;11(5):327–336. - PubMed
-
- Ito Y. RUNX genes in development and cancer: regulation of viral gene expression and the discovery of RUNX family genes. Adv Cancer Res. 2008;99:33–76. - PubMed
-
- Kalev-Zylinska ML, Horsfield JA, Flores MV, et al. Runx1 is required for zebrafish blood and vessel development and expression of a human RUNX1-CBF2T1 transgene advances a model for studies of leukemogenesis. Development. 2002;129(8):2015–2030. - PubMed
-
- de Bruijn MF, Speck NA. Core-binding factors in hematopoiesis and immune function. Oncogene. 2004;23(24):4238–4248. - PubMed
-
- Watanabe-Okochi N, Kitaura J, Ono R, et al. AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood. 2008;111(8):4297–4308. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
