

Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons
- PMID: 20689558
- PMCID: PMC3131888
- DOI: 10.1038/cdd.2010.92
Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons
Abstract
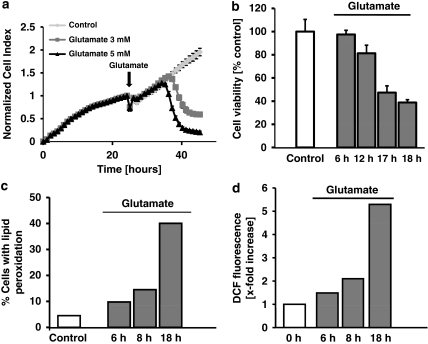
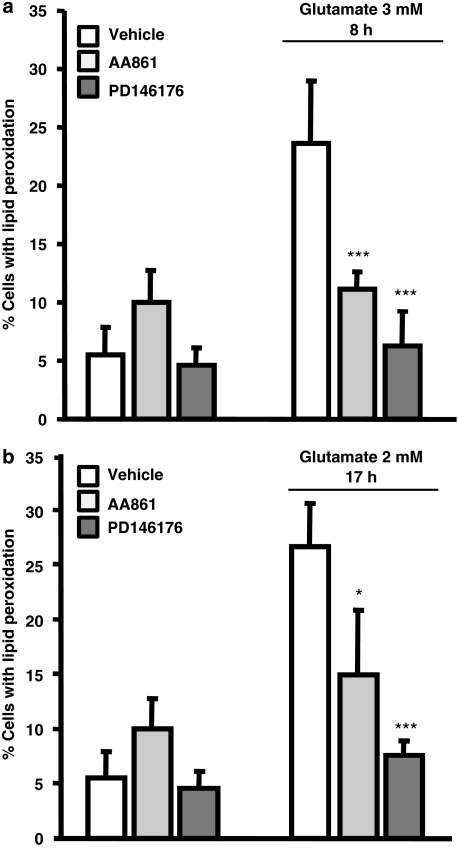
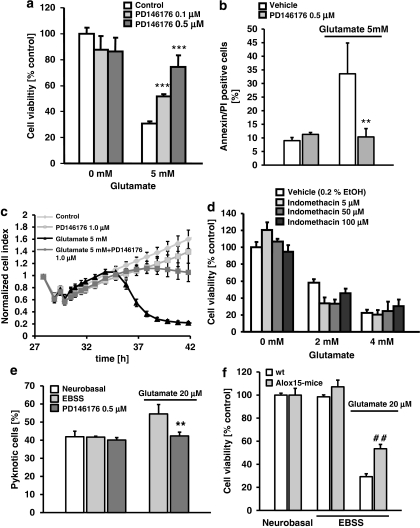
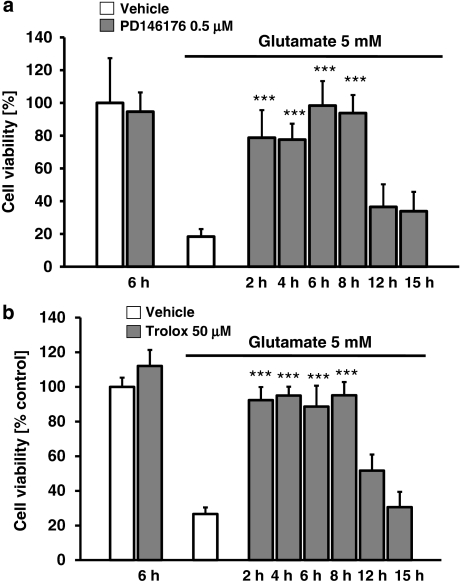
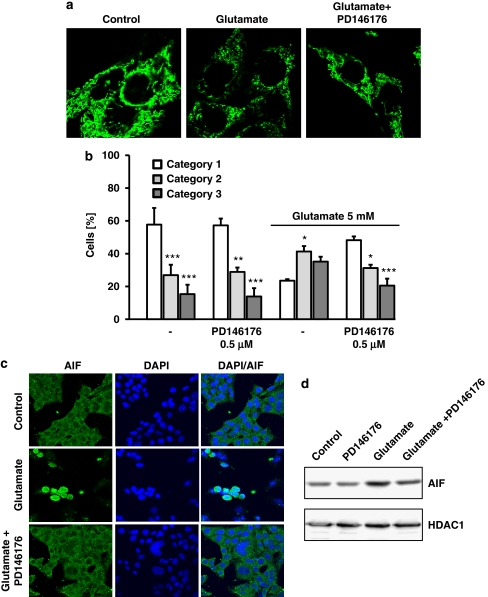
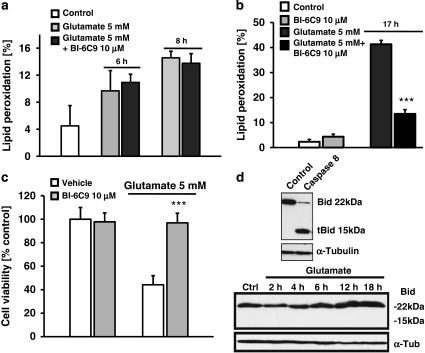
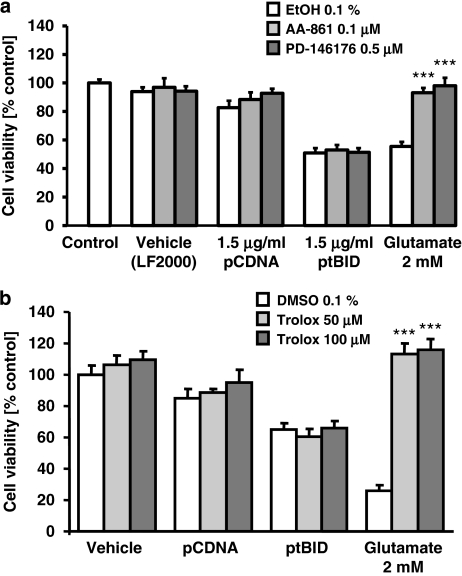
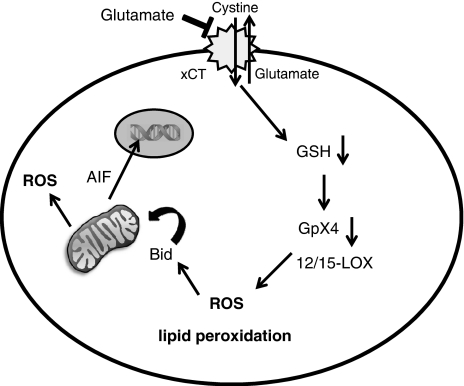
Glutamate toxicity involves increases in intracellular calcium levels and enhanced formation of reactive oxygen species (ROS) causing neuronal dysfunction and death in acute and chronic neurodegenerative disorders. The molecular mechanisms mediating glutamate-induced ROS formation are, however, still poorly defined. Using a model system that lacks glutamate-operated calcium channels, we demonstrate that glutamate-induced acceleration of ROS levels occurs in two steps and is initiated by lipoxygenases (LOXs) and then significantly accelerated through Bid-dependent mitochondrial damage. The Bid-mediated secondary boost of ROS formation downstream of LOX activity further involves mitochondrial fragmentation and release of mitochondrial apoptosis-inducing factor (AIF) to the nucleus. These data imply that the activation of Bid is an essential step in amplifying glutamate-induced formation of lipid peroxides to irreversible mitochondrial damage associated with further enhanced free radical formation and AIF-dependent execution of cell death.
Figures
References
-
- Culmsee C, Krieglstein J. Emerging pharmacotherapeutic strategies for the treatment of ischemic stroke. Drug Discov Today Ther Strateg. 2006;3:621–638.
-
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. - PubMed
-
- Culmsee C, Junker V, Kremers W, Thal S, Plesnila N, Krieglstein J. Combination therapy in ischemic stroke: synergistic neuroprotective effects of memantine and clenbuterol. Stroke. 2004;35:1197–1202. - PubMed
-
- Fisher M, Schaebitz W. An overview of acute stroke therapy: past, present, and future. Arch Intern Med. 2000;160:3196–3206. - PubMed
-
- Matsuda S, Umeda M, Uchida H, Kato H, Araki T. Alterations of oxidative stress markers and apoptosis markers in the striatum after transient focal cerebral ischemia in rats. J Neural Transm. 2009;116:395–404. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
