

Prediction of deleterious non-synonymous SNPs based on protein interaction network and hybrid properties
- PMID: 20689580
- PMCID: PMC2912763
- DOI: 10.1371/journal.pone.0011900
Prediction of deleterious non-synonymous SNPs based on protein interaction network and hybrid properties
Abstract
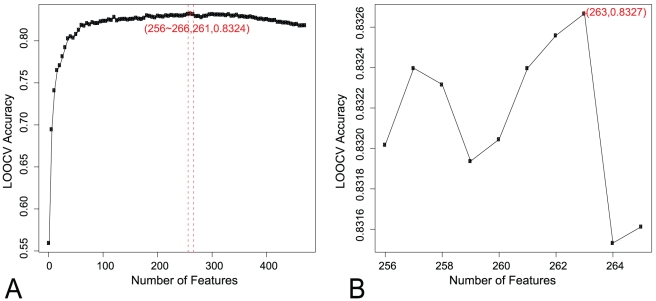
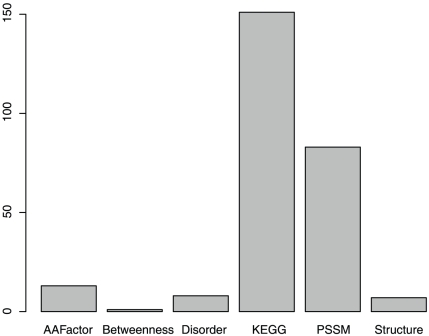
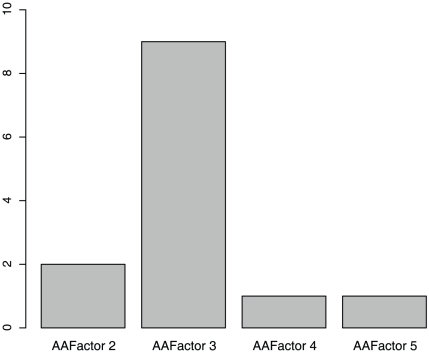
Non-synonymous SNPs (nsSNPs), also known as Single Amino acid Polymorphisms (SAPs) account for the majority of human inherited diseases. It is important to distinguish the deleterious SAPs from neutral ones. Most traditional computational methods to classify SAPs are based on sequential or structural features. However, these features cannot fully explain the association between a SAP and the observed pathophysiological phenotype. We believe the better rationale for deleterious SAP prediction should be: If a SAP lies in the protein with important functions and it can change the protein sequence and structure severely, it is more likely related to disease. So we established a method to predict deleterious SAPs based on both protein interaction network and traditional hybrid properties. Each SAP is represented by 472 features that include sequential features, structural features and network features. Maximum Relevance Minimum Redundancy (mRMR) method and Incremental Feature Selection (IFS) were applied to obtain the optimal feature set and the prediction model was Nearest Neighbor Algorithm (NNA). In jackknife cross-validation, 83.27% of SAPs were correctly predicted when the optimized 263 features were used. The optimized predictor with 263 features was also tested in an independent dataset and the accuracy was still 80.00%. In contrast, SIFT, a widely used predictor of deleterious SAPs based on sequential features, has a prediction accuracy of 71.05% on the same dataset. In our study, network features were found to be most important for accurate prediction and can significantly improve the prediction performance. Our results suggest that the protein interaction context could provide important clues to help better illustrate SAP's functional association. This research will facilitate the post genome-wide association studies.
Conflict of interest statement
Figures
Similar articles
-
SySAP: a system-level predictor of deleterious single amino acid polymorphisms.Protein Cell. 2012 Jan;3(1):38-43. doi: 10.1007/s13238-011-1130-2. Epub 2011 Dec 19. Protein Cell. 2012. PMID: 22183811 Free PMC article.
-
Identification of deleterious non-synonymous single nucleotide polymorphisms using sequence-derived information.BMC Bioinformatics. 2008 Jun 27;9:297. doi: 10.1186/1471-2105-9-297. BMC Bioinformatics. 2008. PMID: 18588693 Free PMC article.
-
Predicting deleterious non-synonymous single nucleotide polymorphisms in signal peptides based on hybrid sequence attributes.Comput Biol Chem. 2012 Feb;36:31-5. doi: 10.1016/j.compbiolchem.2011.12.001. Epub 2011 Dec 30. Comput Biol Chem. 2012. PMID: 22277674
-
Comparison and integration of computational methods for deleterious synonymous mutation prediction.Brief Bioinform. 2020 May 21;21(3):970-981. doi: 10.1093/bib/bbz047. Brief Bioinform. 2020. PMID: 31157880 Review.
-
Computational prediction of the effects of non-synonymous single nucleotide polymorphisms in human DNA repair genes.Neuroscience. 2007 Apr 14;145(4):1273-9. doi: 10.1016/j.neuroscience.2006.09.004. Epub 2006 Oct 19. Neuroscience. 2007. PMID: 17055652 Review.
Cited by
-
Improved classification of lung cancer tumors based on structural and physicochemical properties of proteins using data mining models.PLoS One. 2013;8(3):e58772. doi: 10.1371/journal.pone.0058772. Epub 2013 Mar 7. PLoS One. 2013. PMID: 23505559 Free PMC article.
-
Single nucleotide polymorphisms in microRNA binding sites of oncogenes: implications in cancer and pharmacogenomics.OMICS. 2014 Feb;18(2):142-54. doi: 10.1089/omi.2013.0098. Epub 2013 Nov 28. OMICS. 2014. PMID: 24286505 Free PMC article.
-
Systems pharmacology: network analysis to identify multiscale mechanisms of drug action.Annu Rev Pharmacol Toxicol. 2012;52:505-21. doi: 10.1146/annurev-pharmtox-010611-134520. Annu Rev Pharmacol Toxicol. 2012. PMID: 22235860 Free PMC article. Review.
-
SySAP: a system-level predictor of deleterious single amino acid polymorphisms.Protein Cell. 2012 Jan;3(1):38-43. doi: 10.1007/s13238-011-1130-2. Epub 2011 Dec 19. Protein Cell. 2012. PMID: 22183811 Free PMC article.
-
IDRMutPred: predicting disease-associated germline nonsynonymous single nucleotide variants (nsSNVs) in intrinsically disordered regions.Bioinformatics. 2020 Dec 22;36(20):4977-4983. doi: 10.1093/bioinformatics/btaa618. Bioinformatics. 2020. PMID: 32756939 Free PMC article.
References
-
- Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998;8:1229–1231. - PubMed
-
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
