

Massive turnover of functional sequence in human and other mammalian genomes
- PMID: 20693480
- PMCID: PMC2945182
- DOI: 10.1101/gr.108795.110
Massive turnover of functional sequence in human and other mammalian genomes
Abstract
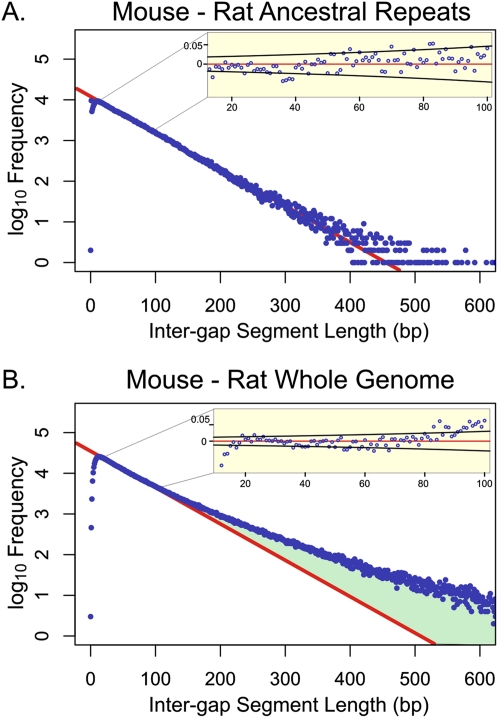
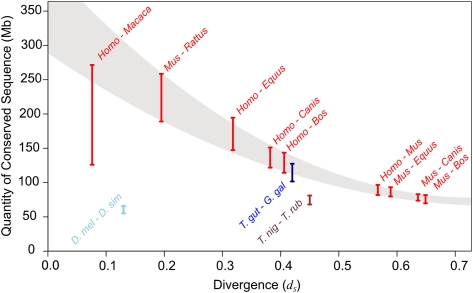
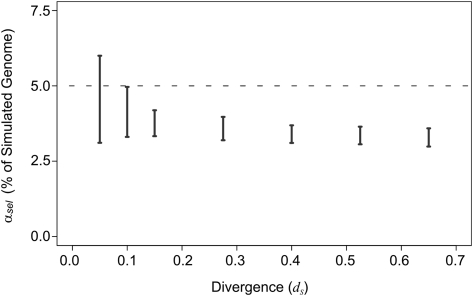
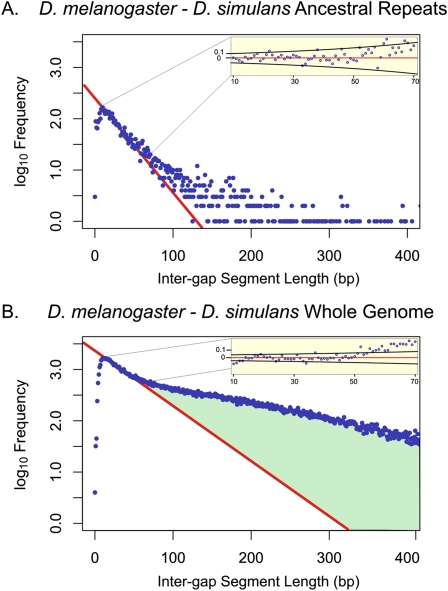
Despite the availability of dozens of animal genome sequences, two key questions remain unanswered: First, what fraction of any species' genome confers biological function, and second, are apparent differences in organismal complexity reflected in an objective measure of genomic complexity? Here, we address both questions by applying, across the mammalian phylogeny, an evolutionary model that estimates the amount of functional DNA that is shared between two species' genomes. Our main findings are, first, that as the divergence between mammalian species increases, the predicted amount of pairwise shared functional sequence drops off dramatically. We show by simulations that this is not an artifact of the method, but rather indicates that functional (and mostly noncoding) sequence is turning over at a very high rate. We estimate that between 200 and 300 Mb (∼6.5%-10%) of the human genome is under functional constraint, which includes five to eight times as many constrained noncoding bases than bases that code for protein. In contrast, in D. melanogaster we estimate only 56-66 Mb to be constrained, implying a ratio of noncoding to coding constrained bases of about 2. This suggests that, rather than genome size or protein-coding gene complement, it is the number of functional bases that might best mirror our naïve preconceptions of organismal complexity.
Figures
Similar articles
-
8.2% of the Human genome is constrained: variation in rates of turnover across functional element classes in the human lineage.PLoS Genet. 2014 Jul 24;10(7):e1004525. doi: 10.1371/journal.pgen.1004525. eCollection 2014 Jul. PLoS Genet. 2014. PMID: 25057982 Free PMC article.
-
Rapid turnover of functional sequence in human and other genomes.Annu Rev Genomics Hum Genet. 2011;12:275-99. doi: 10.1146/annurev-genom-090810-183115. Annu Rev Genomics Hum Genet. 2011. PMID: 21721940 Review.
-
Evolutionary history of mammalian transposons determined by genome-wide defragmentation.PLoS Comput Biol. 2007 Jul;3(7):e137. doi: 10.1371/journal.pcbi.0030137. PLoS Comput Biol. 2007. PMID: 17630829 Free PMC article.
-
The role of transposable elements in the evolution of non-mammalian vertebrates and invertebrates.Genome Biol. 2010;11(6):R59. doi: 10.1186/gb-2010-11-6-r59. Epub 2010 Jun 2. Genome Biol. 2010. PMID: 20525173 Free PMC article.
-
What fraction of the human genome is functional?Genome Res. 2011 Nov;21(11):1769-76. doi: 10.1101/gr.116814.110. Epub 2011 Aug 29. Genome Res. 2011. PMID: 21875934 Free PMC article. Review.
Cited by
-
ChIP-ping the branches of the tree: functional genomics and the evolution of eukaryotic gene regulation.Brief Funct Genomics. 2018 Mar 1;17(2):116-137. doi: 10.1093/bfgp/ely004. Brief Funct Genomics. 2018. PMID: 29529131 Free PMC article. Review.
-
8.2% of the Human genome is constrained: variation in rates of turnover across functional element classes in the human lineage.PLoS Genet. 2014 Jul 24;10(7):e1004525. doi: 10.1371/journal.pgen.1004525. eCollection 2014 Jul. PLoS Genet. 2014. PMID: 25057982 Free PMC article.
-
SuRFing the genomics wave: an R package for prioritising SNPs by functionality.Genome Med. 2014 Oct 14;6(10):79. doi: 10.1186/s13073-014-0079-1. eCollection 2014. Genome Med. 2014. PMID: 25400697 Free PMC article.
-
A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data.Sci Rep. 2015 May 27;5:10576. doi: 10.1038/srep10576. Sci Rep. 2015. PMID: 26015273 Free PMC article.
-
29 mammalian genomes reveal novel exaptations of mobile elements for likely regulatory functions in the human genome.PLoS One. 2012;7(8):e43128. doi: 10.1371/journal.pone.0043128. Epub 2012 Aug 27. PLoS One. 2012. PMID: 22952639 Free PMC article.
References
-
- Andolfatto P 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437: 1149–1152 - PubMed
-
- Batzer MA, Deininger PL 2002. Alu repeats and human genomic diversity. Nat Rev Genet 3: 370–379 - PubMed
-
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D 2004. Ultraconserved elements in the human genome. Science 304: 1321–1325 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous