

A Non-Orthogonal Block-Localized Effective Hamiltonian Approach for Chemical and Enzymatic Reactions
- PMID: 20694172
- PMCID: PMC2914346
- DOI: 10.1021/ct1001686
A Non-Orthogonal Block-Localized Effective Hamiltonian Approach for Chemical and Enzymatic Reactions
Abstract
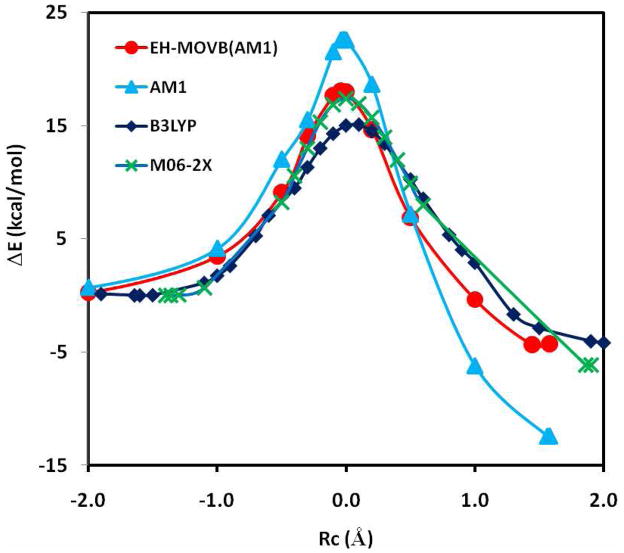
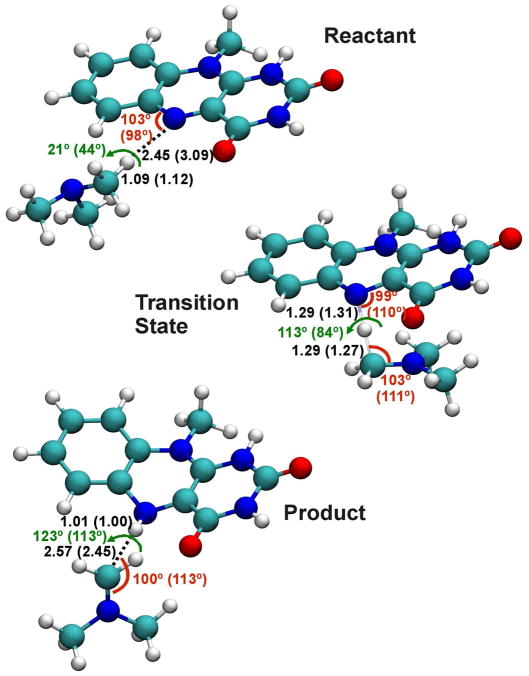
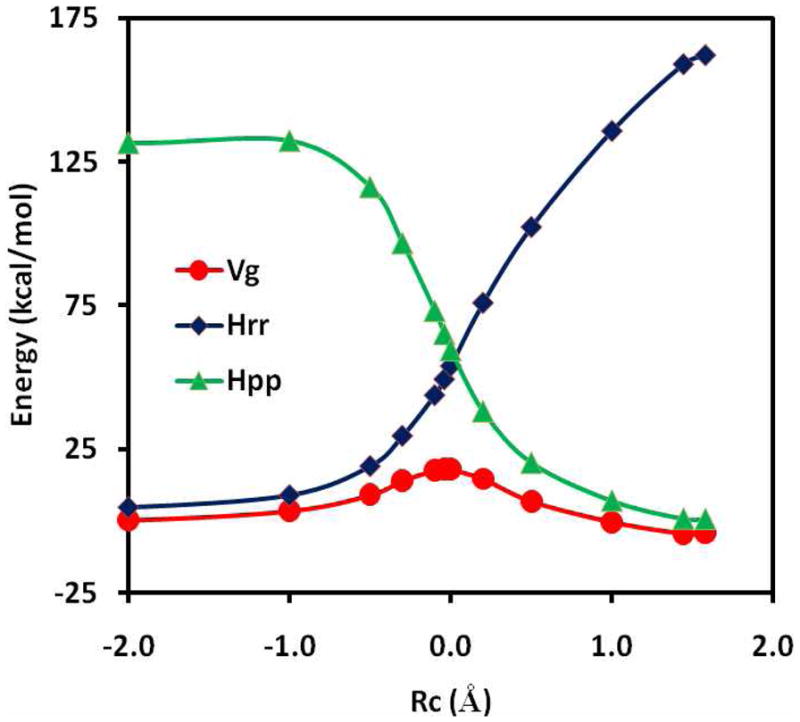
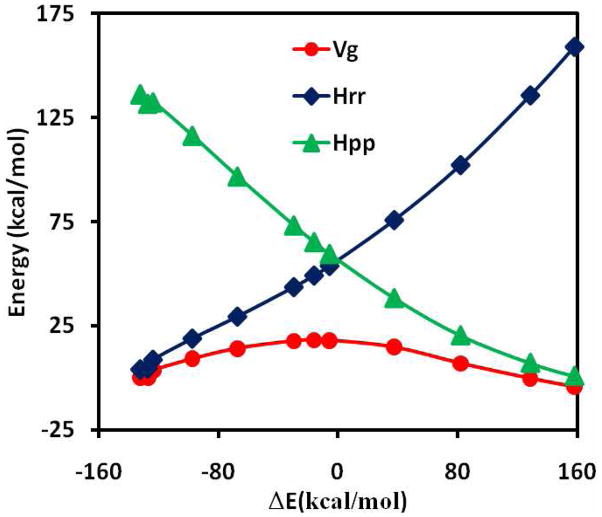
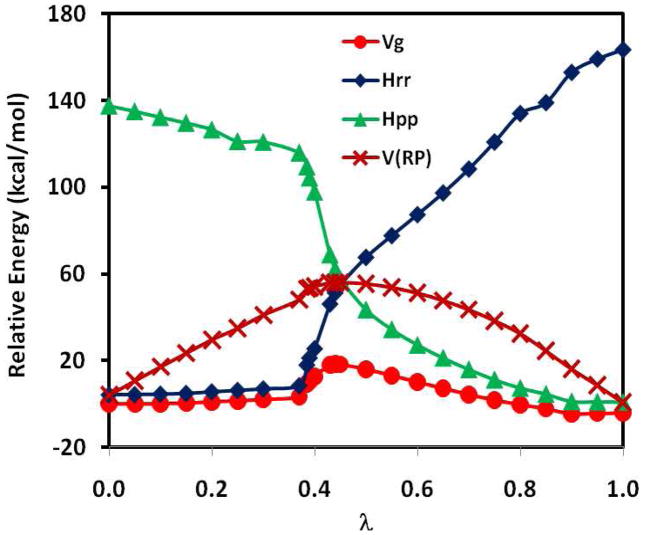
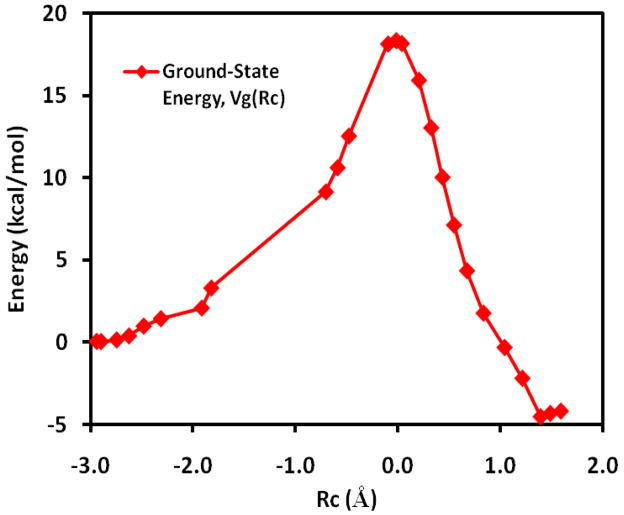
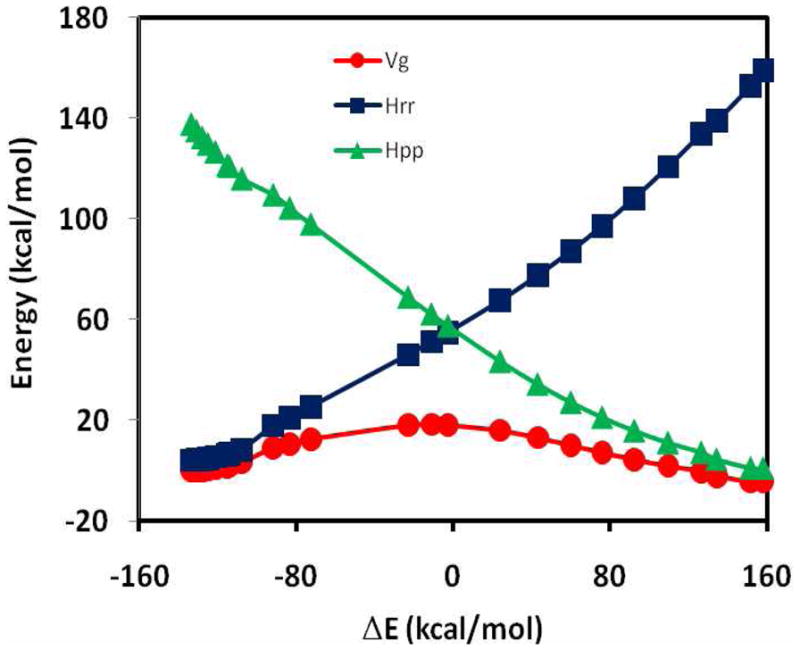
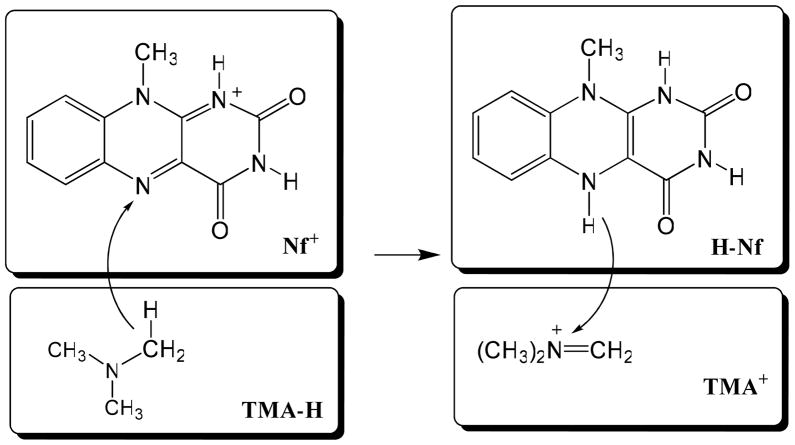
The effective Hamiltonian-molecular orbital and valence bond (EH-MOVB) method based on non-orthogonal block-localized fragment orbitals has been implemented into the program CHARMM for molecular dynamics simulations of chemical and enzymatic reactions, making use of semiempirical quantum mechanical models. Building upon ab initio MOVB theory, we make use of two parameters in the EH-MOVB method to fit the barrier height and the relative energy between the reactant and product state for a given chemical reaction to be in agreement with experiment or high-level ab initio or density functional results. Consequently, the EH-MOVB method provides a highly accurate and computationally efficient QM/MM model for dynamics simulation of chemical reactions in solution. The EH-MOVB method is illustrated by examination of the potential energy surface of the hydride transfer reaction from trimethylamine to a flavin cofactor model in the gas phase. In the present study, we employed the semiempirical AM1 model, which yields a reaction barrier that is more than 5 kcal/mol too high. We use a parameter calibration procedure for the EH-MOVB method similar to that employed to adjust the results of semiempirical and empirical models. Thus, the relative energy of these two diabatic states can be shifted to reproduce the experimental energy of reaction, and the barrier height is optimized to reproduce the desired (accurate) value by adding a constant to the off-diagonal matrix element. The present EH-MOVB method offers a viable approach to characterizing solvent and protein-reorganization effects in the realm of combined QM/MM simulations.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources