

Research into Specific Modulators of Vascular Sex Hormone Receptors in the Management of Postmenopausal Cardiovascular Disease
- PMID: 20694192
- PMCID: PMC2915874
- DOI: 10.2174/157340209789587717
Research into Specific Modulators of Vascular Sex Hormone Receptors in the Management of Postmenopausal Cardiovascular Disease
Abstract
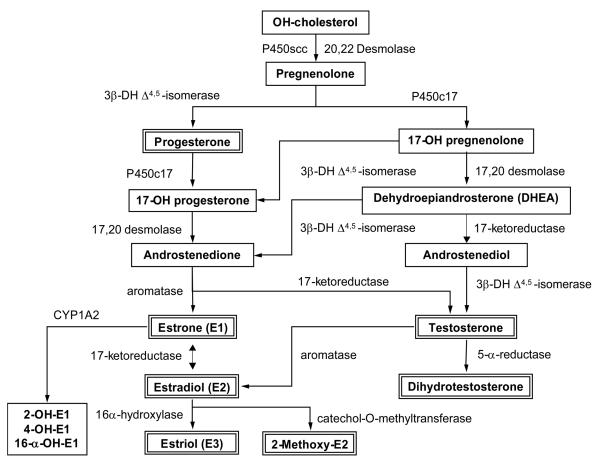
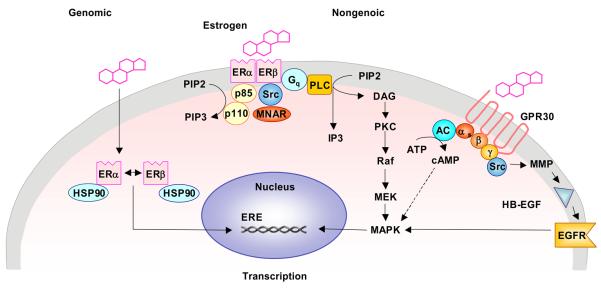
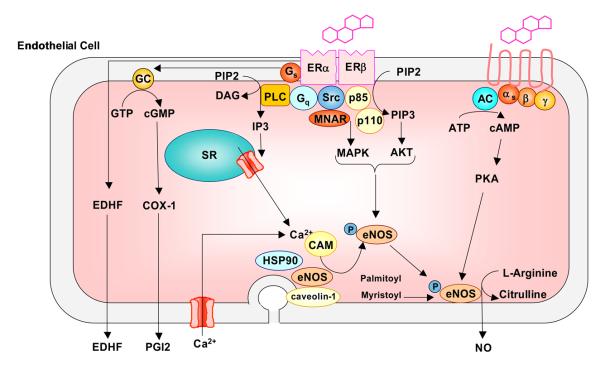
Cardiovascular disease (CVD) is more common in men and postmenopausal women than premenopausal women, suggesting vascular benefits of female sex hormones. Studies on the vasculature have identified estrogen receptors ERα, ERβ and a novel estrogen binding membrane protein GPR30, that mediate genomic and/or non-genomic effects. Estrogen promotes endothelium-dependent relaxation by inducing the production/activity of nitric oxide, prostacyclin, and hyperpolarizing factor, and inhibits the mechanisms of vascular smooth muscle contraction including [Ca(2+)](i), protein kinase C, Rho kinase and mitogen-activated protein kinase. Additional effects of estrogen on the cytoskeleton, matrix metalloproteinases and inflammatory factors contribute to vascular remodeling. However, the experimental evidence did not translate into vascular benefits of menopausal hormone therapy (MHT), and the HERS, HERS-II and WHI clinical trials demonstrated adverse cardiovascular events. The discrepancy has been partly related to delayed MHT and potential changes in the vascular ER amount, integrity, affinity, and downstream signaling pathways due to the subjects' age and preexisting CVD. The adverse vascular effects of MHT also highlighted the need of specific modulators of vascular sex hormone receptors. The effectiveness of MHT can be improved by delineating the differences in phramcokinetics and pharmacodynamics of natural, synthetic, and conjugated equine estrogens. Estriol, "hormone bioidenticals" and phytoestrogens are potential estradiol substitutes. The benefits of low dose MHT, and transdermal or vaginal estrogens over oral preparations are being evaluated. Specific ER modulators (SERMs) and ER agonists are being developed to maximize the effects on vascular ERs. Also, the effects of estrogen are being examined in the context of the whole body hormonal environment and the levels of progesterone and androgens. Thus, the experimental vascular benefits of estrogen can be translated to the outcome of MHT in postmenopausal CVD, as more specific modulators of sex hormone receptors become available and are used at the right dose, route of administration and timing, depending on the subject's age and preexisting cardiovascular condition.
Figures


Similar articles
-
Estrogen, vascular estrogen receptor and hormone therapy in postmenopausal vascular disease.Biochem Pharmacol. 2013 Dec 15;86(12):1627-42. doi: 10.1016/j.bcp.2013.09.024. Epub 2013 Oct 4. Biochem Pharmacol. 2013. PMID: 24099797 Free PMC article.
-
Experimental benefits of sex hormones on vascular function and the outcome of hormone therapy in cardiovascular disease.Curr Cardiol Rev. 2008 Nov;4(4):309-22. doi: 10.2174/157340308786349462. Curr Cardiol Rev. 2008. PMID: 20066139 Free PMC article.
-
Modulators of vascular sex hormone receptors and their effects in estrogen-deficiency states associated with menopause.Recent Pat Cardiovasc Drug Discov. 2008 Nov;3(3):165-86. doi: 10.2174/157489008786263970. Recent Pat Cardiovasc Drug Discov. 2008. PMID: 18991792 Review.
-
Vascular effects of estrogenic menopausal hormone therapy.Rev Recent Clin Trials. 2012 Feb;7(1):47-70. doi: 10.2174/157488712799363253. Rev Recent Clin Trials. 2012. PMID: 21864249 Free PMC article. Review.
-
Potential approaches to enhance the effects of estrogen on senescent blood vessels and postmenopausal cardiovascular disease.Cardiovasc Hematol Agents Med Chem. 2010 Jan;8(1):29-46. doi: 10.2174/187152510790796156. Cardiovasc Hematol Agents Med Chem. 2010. PMID: 20210774 Free PMC article. Review.
Cited by
-
Cyclic guanosine monophosphate and 10-year change in left ventricular mass: the Multi-Ethnic Study of Atherosclerosis (MESA).Biomarkers. 2021 Jun;26(4):309-317. doi: 10.1080/1354750X.2021.1893811. Epub 2021 Mar 9. Biomarkers. 2021. PMID: 33715578 Free PMC article.
-
Effect of sex, menstrual cycle phase, and monophasic oral contraceptive pill use on local and central arterial stiffness in young adults.Am J Physiol Heart Circ Physiol. 2018 Aug 1;315(2):H357-H365. doi: 10.1152/ajpheart.00039.2018. Epub 2018 Apr 20. Am J Physiol Heart Circ Physiol. 2018. PMID: 29677465 Free PMC article.
-
Network Pharmacology-Based Prediction of Active Ingredients and Potential Targets of ShengDiHuang Decoction for Treatment of Dysfunctional Uterine Bleeding.Evid Based Complement Alternat Med. 2020 May 11;2020:7370304. doi: 10.1155/2020/7370304. eCollection 2020. Evid Based Complement Alternat Med. 2020. PMID: 32454870 Free PMC article.
-
Assessing the Anti-inflammatory Mechanism of Reduning Injection by Network Pharmacology.Biomed Res Int. 2020 Dec 16;2020:6134098. doi: 10.1155/2020/6134098. eCollection 2020. Biomed Res Int. 2020. PMID: 33381562 Free PMC article.
-
Male-female comparison of vasomotor effects of circulating hormones in human intracranial arteries.J Headache Pain. 2024 Dec 11;25(1):216. doi: 10.1186/s10194-024-01933-w. J Headache Pain. 2024. PMID: 39663536 Free PMC article.
References
-
- Adams M, Thomas RC, Golden DL, Wagner JD, Williams JK. Mehtoxyprogesterone acetate antagonizes inhibitory effects of conjugated equine estrogens on coronary artery artherosclerosis. Arteriosclerosis, Thrombosis and Vascular Biology. 1997;17:217–221. - PubMed
-
- Adashi EY, Jones PB, Hsueh AJ. Synergistic effect of glucocorticoids on the stimulation of progesterone production by follicle-stimulating hormone in cultured rat granulosa cells. Endocrinology. 1981;109(6):1888–94. - PubMed
-
- Adashi EY. The climacteric ovary as a functional gonadotropin-driven androgen-producing gland. Fertil Steril. 1994;62(1):20–7. - PubMed
-
- Allolio B, Arlt W. DHEA treatment: myth or reality? Trends Endocrinol Metab. 2002;13(7):288–94. - PubMed
-
- Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA. 2004;291(14):1701–12. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous