

Molecular pathology of human prion disease
- PMID: 20694796
- PMCID: PMC3015177
- DOI: 10.1007/s00401-010-0735-5
Molecular pathology of human prion disease
Abstract
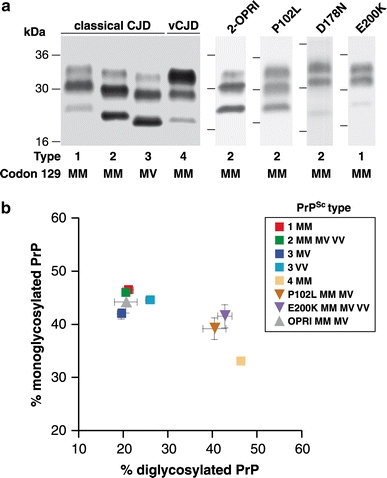
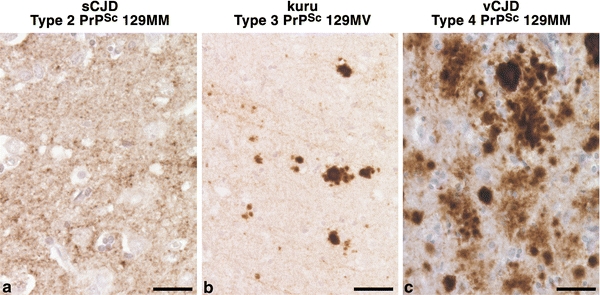
Human prion diseases are associated with a range of clinical presentations and are classified by both clinicopathological syndrome and aetiology with sub-classification according to molecular criteria. Considerable experimental evidence suggests that phenotypic diversity in human prion disease relates in significant part to the existence of distinct human prion strains encoded by abnormal PrP isoforms with differing physicochemical properties. To date, however, the conformational repertoire of pathological isoforms of wild-type human PrP and the various forms of mutant human PrP has not been fully defined. Efforts to produce a unified international classification of human prion disease are still ongoing. The ability of genetic background to influence prion strain selection together with knowledge of numerous other factors that may influence clinical and neuropathological presentation strongly emphasises the requirement to identify distinct human prion strains in appropriate transgenic models, where host genetic variability and other modifiers of phenotype are removed. Defining how many human prion strains exist allied with transgenic modelling of potentially zoonotic prion strains will inform on how many human infections may have an animal origin. Understanding these relationships will have direct translation to protecting public health.
Figures
References
-
- Alpers MP (1987) Epidemiology and clinical aspects of kuru. In: Prusiner SB, McKinley MP (eds) Prions: novel infectious pathogens causing scrapie and Creutzfeldt-Jakob disease. Academic Press, San Diego, pp 451–465
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
