

Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology
- PMID: 20696699
- PMCID: PMC2931174
- DOI: 10.1084/jem.20092749
Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology
Abstract
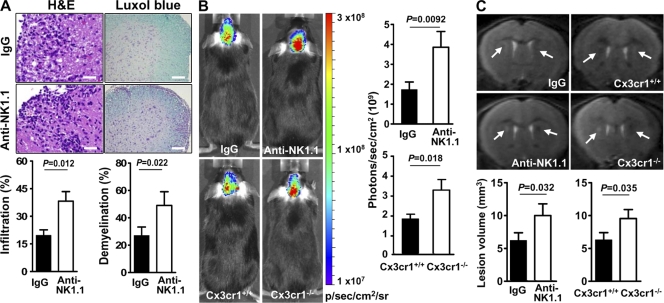
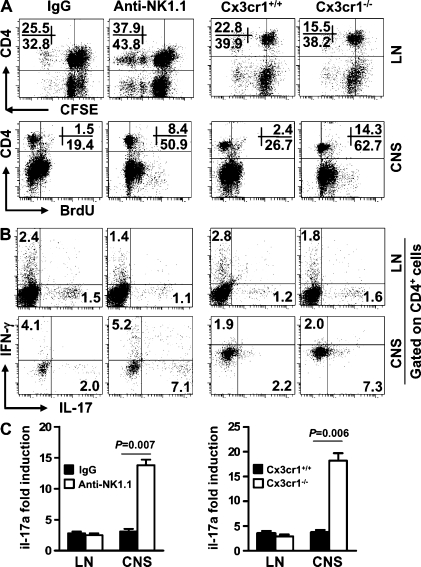
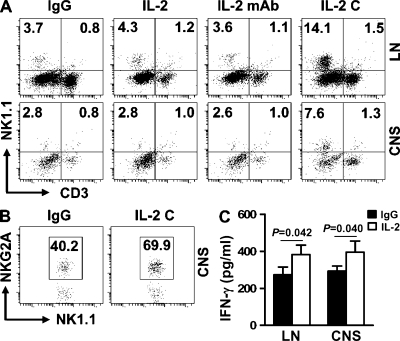
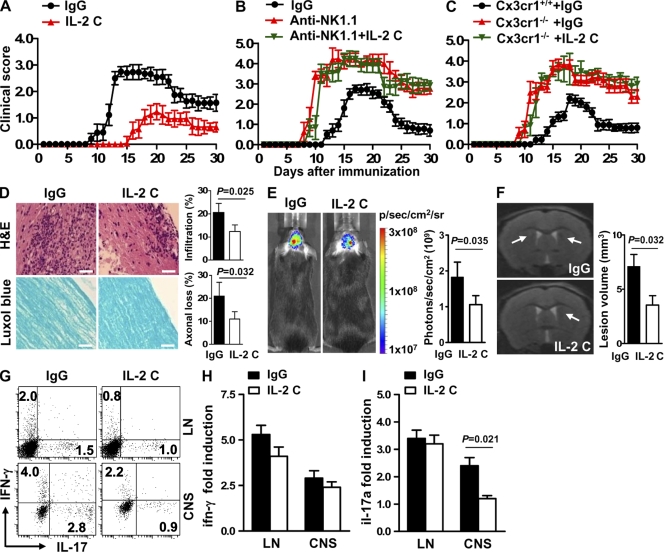
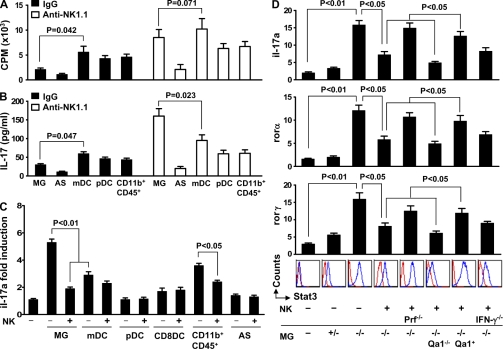
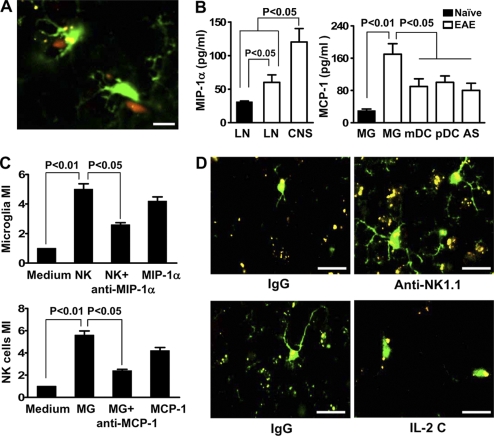
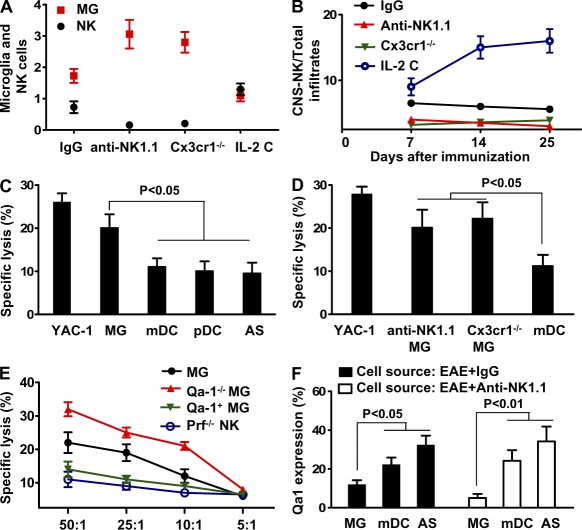
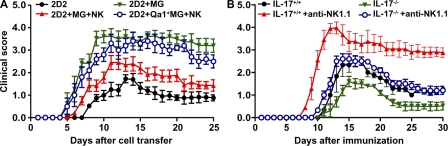
Natural killer (NK) cells of the innate immune system can profoundly impact the development of adaptive immune responses. Inflammatory and autoimmune responses in anatomical locations such as the central nervous system (CNS) differ substantially from those found in peripheral organs. We show in a mouse model of multiple sclerosis that NK cell enrichment results in disease amelioration, whereas selective blockade of NK cell homing to the CNS results in disease exacerbation. Importantly, the effects of NK cells on CNS pathology were dependent on the activity of CNS-resident, but not peripheral, NK cells. This activity of CNS-resident NK cells involved interactions with microglia and suppression of myelin-reactive Th17 cells. Our studies suggest an organ-specific activity of NK cells on the magnitude of CNS inflammation, providing potential new targets for therapeutic intervention.
Figures
References
-
- Abizaid A., Liu Z.W., Andrews Z.B., Shanabrough M., Borok E., Elsworth J.D., Roth R.H., Sleeman M.W., Picciotto M.R., Tschöp M.H., et al. 2006. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Invest. 116:3229–3239 10.1172/JCI29867 - DOI - PMC - PubMed
-
- Assarsson E., Kambayashi T., Schatzle J.D., Cramer S.O., von Bonin A., Jensen P.E., Ljunggren H.G., Chambers B.J. 2004. NK cells stimulate proliferation of T and NK cells through 2B4/CD48 interactions. J. Immunol. 173:174–180 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
