

Viral hijacking of the host ubiquitin system to evade interferon responses
- PMID: 20699190
- PMCID: PMC2939720
- DOI: 10.1016/j.mib.2010.05.012
Viral hijacking of the host ubiquitin system to evade interferon responses
Abstract
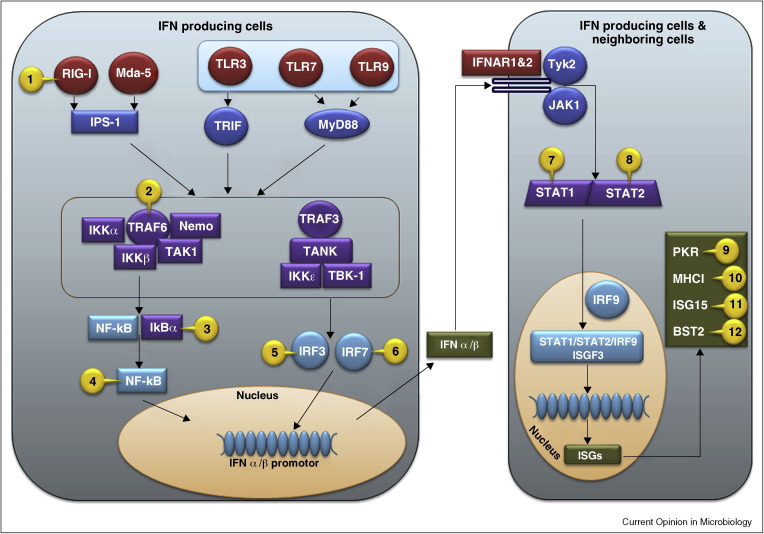
The post-translational attachment of ubiquitin or ubiquitin-like modifiers (ULMs) to proteins regulates many cellular processes including the generation of innate and adaptive immune responses to pathogens. Vice versa, pathogens counteract immune defense by inhibiting or redirecting the ubiquitination machinery of the host. A common immune evasion strategy is for viruses to target host immunoproteins for proteasomal or lysosomal degradation by employing viral or host ubiquitin ligases. By degrading key host adaptor and signaling molecules, viruses thus disable multiple immune response pathways including the production of and response to interferons as well as other innate host defense mechanisms. Recent work further revealed that viruses inhibit the ligation of ubiquitin or ULMs or remove ubiquitin from host cell proteins. Thus, viruses succeed in either stabilizing negative regulators of innate immune signaling or thwart host cell proteins that are activated by ubiquitin or ULM-modification.
Copyright 2010 Elsevier Ltd. All rights reserved.
Figures
Comment in
-
The interaction of viruses with host immune defenses.Curr Opin Microbiol. 2010 Aug;13(4):501-2. doi: 10.1016/j.mib.2010.07.001. Epub 2010 Jul 23. Curr Opin Microbiol. 2010. PMID: 20650675 No abstract available.
References
-
- Cadwell K., Coscoy L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science. 2005;309:127–130. - PubMed
-
- Wang X., Herr R.A., Chua W.J., Lybarger L., Wiertz E.J., Hansen T.H. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J Cell Biol. 2007;177:613–624. - PMC - PubMed
-
References 1 and 2 demonstrate that non-lysine residues can be targeted for ubiquitination by E3 ligases.
-
- Randow F., Lehner P.J. Viral avoidance and exploitation of the ubiquitin system. Nat Cell Biol. 2009;11:527–534. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
