

Next generation sequencing in a family with autosomal recessive Kahrizi syndrome (OMIM 612713) reveals a homozygous frameshift mutation in SRD5A3
- PMID: 20700148
- PMCID: PMC3039499
- DOI: 10.1038/ejhg.2010.132
Next generation sequencing in a family with autosomal recessive Kahrizi syndrome (OMIM 612713) reveals a homozygous frameshift mutation in SRD5A3
Abstract
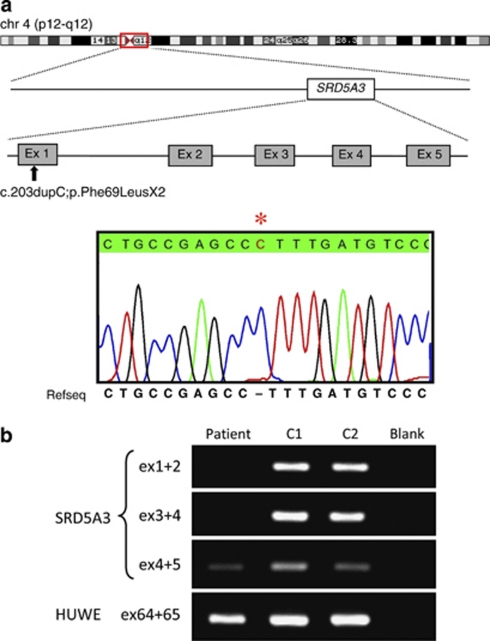
As part of a large-scale, systematic effort to unravel the molecular causes of autosomal recessive mental retardation, we have previously described a novel syndrome consisting of mental retardation, coloboma, cataract and kyphosis (Kahrizi syndrome, OMIM 612713) and mapped the underlying gene to a 10.4-Mb interval near the centromere on chromosome 4. By combining array-based exon enrichment and next generation sequencing, we have now identified a homozygous frameshift mutation (c.203dupC; p.Phe69LeufsX2) in the gene for steroid 5α-reductase type 3 (SRD5A3) as the disease-causing change in this interval. Recent evidence indicates that this enzyme is required for the conversion of polyprenol to dolichol, a step that is essential for N-linked protein glycosylation. Independently, another group has recently observed SRD5A3 mutations in several families with a type 1 congenital disorder of glycosylation (CDG type Ix, OMIM 212067), mental retardation, cerebellar ataxia and eye disorders. Our results show that Kahrizi syndrome and this CDG Ix subtype are allelic disorders, and they illustrate the potential of next-generation sequencing strategies for the elucidation of single gene defects.
Figures
Comment in
-
Normal glycosylation screening does not rule out SRD5A3-CDG.Eur J Hum Genet. 2011 Oct;19(10):1019. doi: 10.1038/ejhg.2010.260. Epub 2011 Jul 13. Eur J Hum Genet. 2011. PMID: 21750573 Free PMC article. No abstract available.
Similar articles
-
Normal glycosylation screening does not rule out SRD5A3-CDG.Eur J Hum Genet. 2011 Oct;19(10):1019. doi: 10.1038/ejhg.2010.260. Epub 2011 Jul 13. Eur J Hum Genet. 2011. PMID: 21750573 Free PMC article. No abstract available.
-
A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism.Brain. 2010 Nov;133(11):3210-20. doi: 10.1093/brain/awq261. Epub 2010 Sep 17. Brain. 2010. PMID: 20852264 Free PMC article.
-
SRD5A3 defective congenital disorder of glycosylation: clinical utility gene card.Eur J Hum Genet. 2020 Sep;28(9):1297-1300. doi: 10.1038/s41431-020-0647-3. Epub 2020 May 18. Eur J Hum Genet. 2020. PMID: 32424323 Free PMC article. No abstract available.
-
Review of SRD5A3 Disease-Causing Sequence Variants and Ocular Findings in Steroid 5α-Reductase Type 3 Congenital Disorder of Glycosylation, and a Detailed New Case.Folia Biol (Praha). 2019;65(3):134-141. doi: 10.14712/fb2019065030134. Folia Biol (Praha). 2019. PMID: 31638560 Review.
-
SRD5A3-CDG: A Patient with a Novel Variant and Brain Neoplasm.J Coll Physicians Surg Pak. 2022 Dec;32(12):SS221-SS226. doi: 10.29271/jcpsp.2022.Supp0.SS221. J Coll Physicians Surg Pak. 2022. PMID: 36597345 Review.
Cited by
-
Cohen Syndrome-Associated Cataract Is Explained by VPS13B Functions in Lens Homeostasis and Is Modified by Additional Genetic Factors.Invest Ophthalmol Vis Sci. 2020 Sep 1;61(11):18. doi: 10.1167/iovs.61.11.18. Invest Ophthalmol Vis Sci. 2020. PMID: 32915983 Free PMC article.
-
Normal glycosylation screening does not rule out SRD5A3-CDG.Eur J Hum Genet. 2011 Oct;19(10):1019. doi: 10.1038/ejhg.2010.260. Epub 2011 Jul 13. Eur J Hum Genet. 2011. PMID: 21750573 Free PMC article. No abstract available.
-
Life with too much polyprenol: polyprenol reductase deficiency.Mol Genet Metab. 2012 Apr;105(4):642-51. doi: 10.1016/j.ymgme.2011.12.017. Epub 2011 Dec 29. Mol Genet Metab. 2012. PMID: 22304929 Free PMC article.
-
Adult phenotype and further phenotypic variability in SRD5A3-CDG.BMC Med Genet. 2014 Jan 16;15:10. doi: 10.1186/1471-2350-15-10. BMC Med Genet. 2014. PMID: 24433453 Free PMC article.
-
Two Argentinean Siblings with CDG-Ix: A Novel Type of Congenital Disorder of Glycosylation?JIMD Rep. 2011;1:65-72. doi: 10.1007/8904_2011_18. Epub 2011 Jun 22. JIMD Rep. 2011. PMID: 23430830 Free PMC article.
References
-
- Morava E, Wosik H, Karteszi J, et al. Congenital disorder of glycosylation type Ix: review of clinical spectrum and diagnostic steps. J Inherit Metab Dis. 2008;31:450–456. - PubMed
-
- Al-Gazali L, Hertecant J, Algawi K, et al. A new autosomal recessive syndrome of ocular colobomas, ichthyosis, brain malformations and endocrine abnormalities in an inbred Emirati family. Am J Med Genet A. 2008;146:813–819. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
