

doi: 10.1186/gb-2010-11-8-r83.
Epub 2010 Aug 11.
Cloud-scale RNA-sequencing differential expression analysis with Myrna
Affiliations
- PMID: 20701754
- PMCID: PMC2945785
- DOI: 10.1186/gb-2010-11-8-r83
Item in Clipboard
Cloud-scale RNA-sequencing differential expression analysis with Myrna
Genome Biol.
2010.
Abstract
As sequencing throughput approaches dozens of gigabases per day, there is a growing need for efficient software for analysis of transcriptome sequencing (RNA-Seq) data. Myrna is a cloud-computing pipeline for calculating differential gene expression in large RNA-Seq datasets. We apply Myrna to the analysis of publicly available data sets and assess the goodness of fit of standard statistical models. Myrna is available from http://bowtie-bio.sf.net/myrna.
Figures
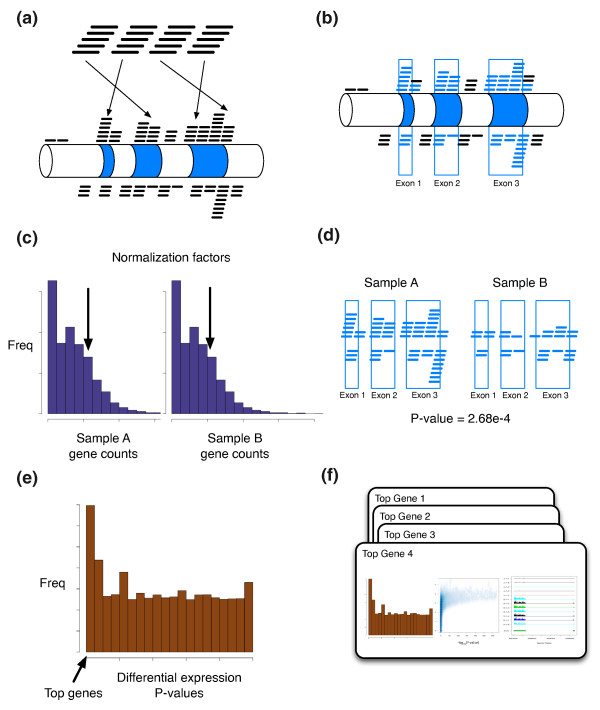
The Myrna pipeline. (a) Reads are aligned to the genome using a parallel version of Bowtie. (b) Reads are aggregated into counts for each genomic feature - for example, for each gene in the annotation files. (c) For each sample a normalization constant is calculated based on a summary of the count distribution. (d) Statistical models are used to calculate differential expression in the R programming language parallelized across multiple processors. (e) Significance summaries such as P-values and gene-specific counts are calculated and returned. (f) Myrna also returns publication ready coverage plots for differentially expressed genes.
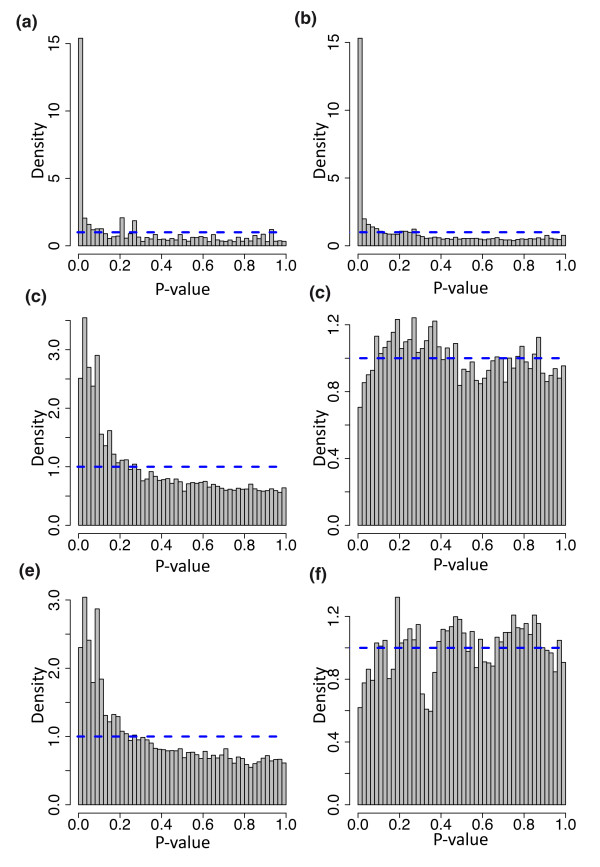
Hapmap results. Histograms of P-values from six different analysis strategies applied to randomly labeled samples. In each case the P-values should be uniformly distributed (blue dotted line) since the labels are randomly assigned. (a) Poisson model, 75th percentile normalization. (b) Poisson model, 75th percentile included as term. (c) Gaussian model, 75th percentile normalization. (d) Gaussian model, 75th percentile included as term. (e) Permutation model, 75th percentile normalization. (f) Permutation model, 75th percentile included as term.
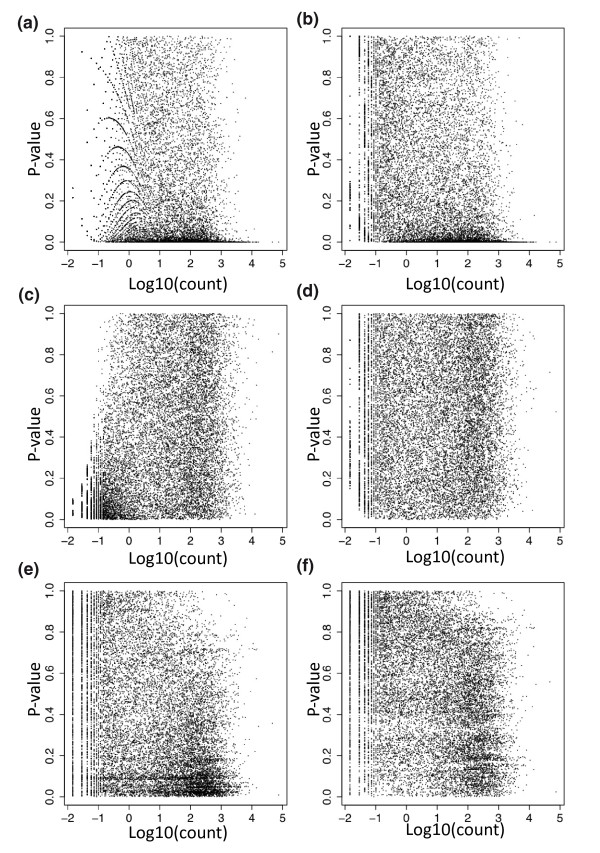
Hapmap P-values versus read depth. A plot of P-value versus the log base 10 of the average count for each gene using the six different analysis strategies applied to randomly labeled samples. In each case the P-values should be uniformly distributed between zero and one. (a) Poisson model, 75th percentile normalization. (b) Poisson model, 75th percentile included as term. (c) Gaussian model, 75th percentile normalization. (d) Gaussian model, 75th percentile included as term. (e) Permutation model, 75th percentile normalization. (f) Permutation model, 75th percentile included as term.
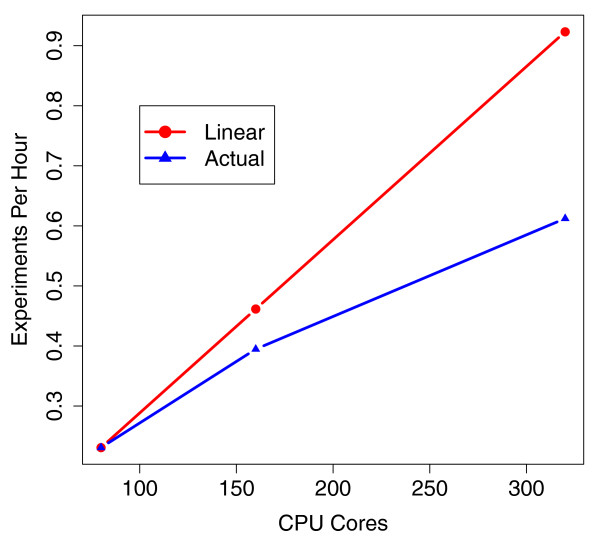
Scalability of Myrna. Number of worker CPU cores allocated from EC2 versus throughput measured in experiments per hour: that is, the reciprocal of the wall clock time required to conduct a whole-human experiment on the 1.1 billion read Pickrell et al. dataset [32]. The line labeled 'linear speedup' traces hypothetical linear speedup relative to the throughput for 80 processor cores.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
