

Receptor for advanced glycation endproducts mediates pro-atherogenic responses to periodontal infection in vascular endothelial cells
- PMID: 20701913
- PMCID: PMC2952730
- DOI: 10.1016/j.atherosclerosis.2010.07.011
Receptor for advanced glycation endproducts mediates pro-atherogenic responses to periodontal infection in vascular endothelial cells
Abstract
Objective: A link between periodontal infections and an increased risk for vascular disease has been demonstrated. Porphyromonas gingivalis, a major periodontal pathogen, localizes in human atherosclerotic plaques, accelerates atherosclerosis in animal models and modulates vascular cell function. The receptor for advanced glycation endproducts (RAGE) regulates vascular inflammation and atherogenesis. We hypothesized that RAGE is involved in P. gingivalis's contribution to pro-atherogenic responses in vascular endothelial cells.
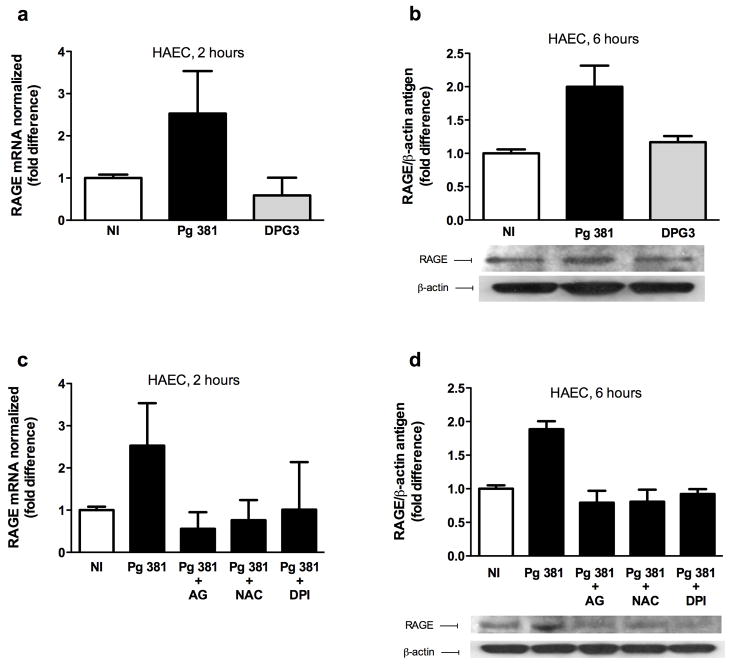
Methods and results: Murine aortic endothelial cells (MAEC) were isolated from wild-type C57BL/6 or RAGE-/- mice and were infected with P. gingivalis strain 381. P. gingivalis 381 infection significantly enhanced expression of RAGE in wild-type MAEC. Levels of pro-atherogenic advanced glycation endproducts (AGEs) and monocyte chemoattractant protein 1 (MCP-1) were significantly increased in wild-type MAEC following P. gingivalis 381 infection, but were unaffected in MAEC from RAGE-/- mice or in MAEC infected with DPG3, a fimbriae-deficient mutant of P. gingivalis 381. Consistent with a role for oxidative stress and an AGE-dependent activation of RAGE in this setting, both antioxidant treatment and AGE blockade significantly suppressed RAGE gene expression and RAGE and MCP-1 protein levels in P. gingivalis 381-infected human aortic endothelial cells (HAEC).
Conclusion: The present findings implicate for the first time the AGE-RAGE axis in the amplification of pro-atherogenic responses triggered by P. gingivalis in vascular endothelial cells.
Copyright © 2010 Elsevier Ireland Ltd. All rights reserved.
Figures




Similar articles
-
Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid β accumulation after Porphyromonas gingivalis infection.J Neurochem. 2021 Aug;158(3):724-736. doi: 10.1111/jnc.15096. Epub 2020 Jun 15. J Neurochem. 2021. PMID: 32441775 Free PMC article.
-
Simvastatin suppresses vascular inflammation and atherosclerosis in ApoE(-/-) mice by downregulating the HMGB1-RAGE axis.Acta Pharmacol Sin. 2013 Jun;34(6):830-6. doi: 10.1038/aps.2013.8. Epub 2013 Apr 8. Acta Pharmacol Sin. 2013. PMID: 23564080 Free PMC article.
-
A murine model of accelerated periodontal disease in diabetes.J Periodontal Res. 1998 Oct;33(7):387-99. doi: 10.1111/j.1600-0765.1998.tb02335.x. J Periodontal Res. 1998. PMID: 9842504
-
Receptor for advanced glycation endproducts and atherosclerosis: From basic mechanisms to clinical implications.Atherosclerosis. 2008 Jan;196(1):9-21. doi: 10.1016/j.atherosclerosis.2007.07.025. Epub 2007 Sep 10. Atherosclerosis. 2008. PMID: 17826783 Review.
-
AGE and their receptor RAGE in systemic autoimmune diseases: an inflammation propagating factor contributing to accelerated atherosclerosis.Autoimmunity. 2009 May;42(4):302-4. doi: 10.1080/08916930902831746. Autoimmunity. 2009. PMID: 19811283 Review.
Cited by
-
Alpha-mangostin attenuation of hyperglycemia-induced ocular hypoperfusion and blood retinal barrier leakage in the early stage of type 2 diabetes rats.Biomed Res Int. 2015;2015:785826. doi: 10.1155/2015/785826. Epub 2015 Apr 9. Biomed Res Int. 2015. PMID: 25950001 Free PMC article.
-
Expression of advanced glycation end products and receptors in gingival tissues of patients with noninsulin-dependent diabetes mellitus-associated periodontitis.J Dent Sci. 2023 Apr;18(2):689-695. doi: 10.1016/j.jds.2022.10.019. Epub 2022 Nov 2. J Dent Sci. 2023. PMID: 37021230 Free PMC article.
-
Diabetes and oral disease: implications for health professionals.Ann N Y Acad Sci. 2012 May;1255:1-15. doi: 10.1111/j.1749-6632.2011.06460.x. Epub 2012 Mar 12. Ann N Y Acad Sci. 2012. PMID: 22409777 Free PMC article.
-
Moxibustion attenuates inflammation in intestinal mucosal by regulating RAGE-mediated TLR4-NF-κBp65 signaling pathway in vivo and in vitro.Am J Transl Res. 2022 Jun 15;14(6):4278-4294. eCollection 2022. Am J Transl Res. 2022. PMID: 35836884 Free PMC article.
-
Phosphatidylserine receptors: enhancers of enveloped virus entry and infection.Virology. 2014 Nov;468-470:565-580. doi: 10.1016/j.virol.2014.09.009. Epub 2014 Sep 29. Virology. 2014. PMID: 25277499 Free PMC article. Review.
References
-
- Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–20. - PubMed
-
- Beck JD, Offenbacher S. The association between periodontal diseases and cardiovascular diseases: a state-of-the-science review. Ann Periodontol. 2001;6:9–15. - PubMed
-
- Kinane DF, Riggio MP, Walker KF, MacKenzie D, Shearer B. Bacteraemia following periodontal procedures. J Clin Periodontol. 2005;32:708–13. - PubMed
-
- Lalla E, Lamster IB, Hofmann MA, et al. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2003;23:1405–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
