

A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5
- PMID: 20702642
- PMCID: PMC2950574
- DOI: 10.1128/JVI.01109-10
A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5
Abstract
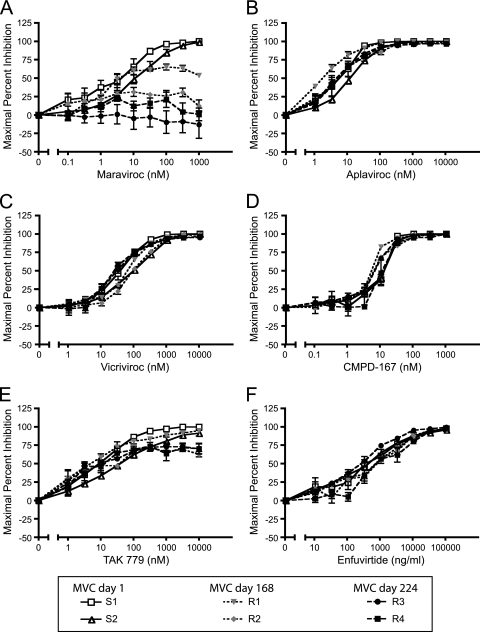
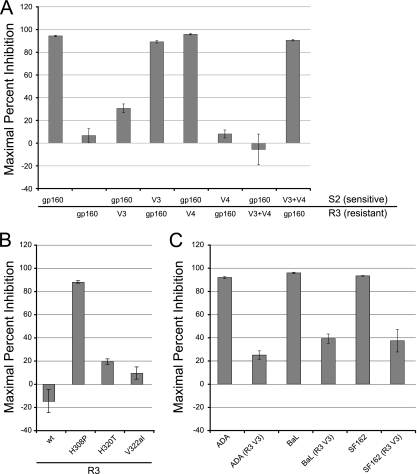
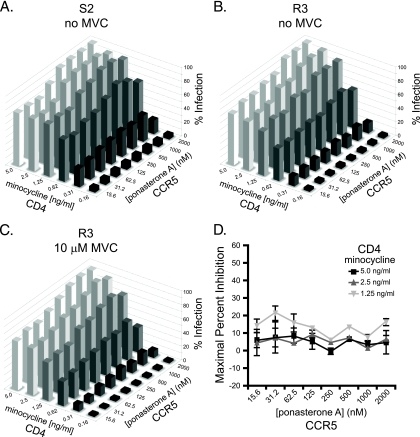
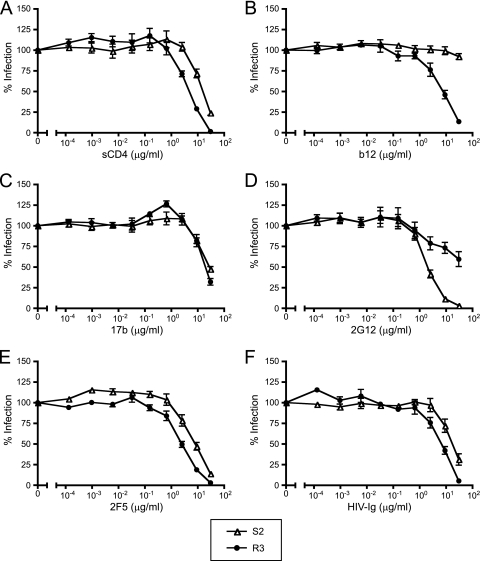
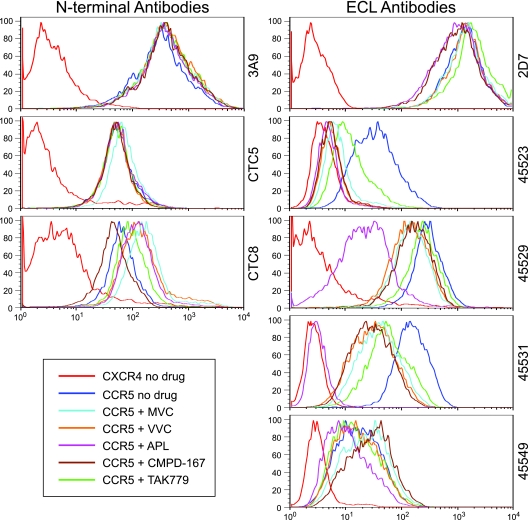
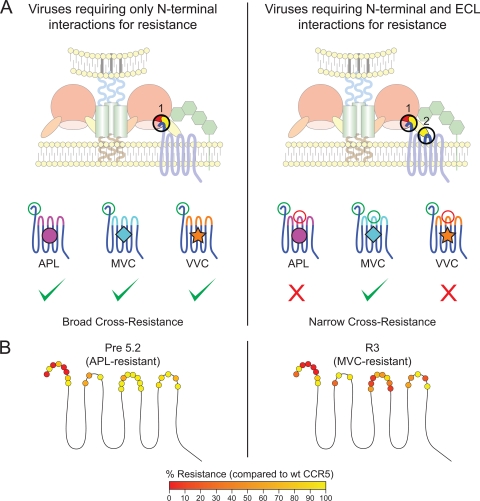
CCR5 antagonists inhibit HIV entry by binding to a coreceptor and inducing changes in the extracellular loops (ECLs) of CCR5. In this study, we analyzed viruses from 11 treatment-experienced patients who experienced virologic failure on treatment regimens containing the CCR5 antagonist maraviroc (MVC). Viruses from one patient developed high-level resistance to MVC during the course of treatment. Although resistance to one CCR5 antagonist is often associated with broad cross-resistance to other agents, these viruses remained sensitive to most other CCR5 antagonists, including vicriviroc and aplaviroc. MVC resistance was dependent upon mutations within the V3 loop of the viral envelope (Env) protein and was modulated by additional mutations in the V4 loop. Deep sequencing of pretreatment plasma viral RNA indicated that resistance appears to have occurred by evolution of drug-bound CCR5 use, despite the presence of viral sequences predictive of CXCR4 use. Envs obtained from this patient before and during MVC treatment were able to infect cells expressing very low CCR5 levels, indicating highly efficient use of a coreceptor. In contrast to previous reports in which CCR5 antagonist-resistant viruses interact predominantly with the N terminus of CCR5, these MVC-resistant Envs were also dependent upon the drug-modified ECLs of CCR5 for entry. Our results suggest a model of CCR5 cross-resistance whereby viruses that predominantly utilize the N terminus are broadly cross-resistant to multiple CCR5 antagonists, whereas viruses that require both the N terminus and antagonist-specific ECL changes demonstrate a narrow cross-resistance profile.
Figures
References
-
- Abebe, A., D. Demissie, J. Goudsmit, M. Brouwer, C. L. Kuiken, G. Pollakis, H. Schuitemaker, A. L. Fontanet, and T. F. Rinke de Wit. 1999. HIV-1 subtype C syncytium- and non-syncytium-inducing phenotypes and coreceptor usage among Ethiopian patients with AIDS. AIDS 13:1305-1311. - PubMed
-
- Atchison, R. E., J. Gosling, F. S. Monteclaro, C. Franci, L. Digilio, I. F. Charo, and M. A. Goldsmith. 1996. Multiple extracellular elements of CCR5 and HIV-1 entry: dissociation from response to chemokines. Science 274:1924-1926. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
